Computer System Validation – CSV
Computer System Validation bezeichnet einen dokumentierten Prozess, der sicherstellt, dass ein computergestütztes System genau das tut, wofür es entwickelt wurde, und zwar auf konsistente und reproduzierbare Weise.
Der Validierungsprozess liefert den dokumentierten Nachweis, dass das System durchgängig seine vordefinierten Anforderungen erfüllt und für seinen bestimmungsgemäßen Gebrauch geeignet ist. Validierungen können in verschiedenen Typen des GxP-regulierten Umfelds verwendet werden:
- Sterilität
- Reinigung
- Methode
- Prozessvalidierung
Warum ist die Computer System Validation wichtig?
Insbesondere für Pharmaunternehmen und Medizinproduktehersteller ist die Computer System Validation relevant, da hier die Transparenz, Qualität sowie Patientensicherheit des Systems bewertet wird. Dabei gilt stets:
- Vermeidung von Gefahren und Risiken für die menschliche Gesundheit und das Leben
- Vermeidung von Produktfehlern und deren Auswirkungen auf die menschliche Gesundheit und das Leben
- Gewährleistung von qualitativ hochwertigen Produkten durch umfangreiche Tests und einen kontrollierten IT-Betrieb
- Minimieren Sie das Risiko von Fehlfunktionen und Ausfällen durch einen kontrollierten IT-Betrieb und transparente IT-Prozesse
Mit Umsetzung der Computer System Validation stellen Sie zudem sicher, dass Sie im Einklang mit nationalen sowie internationalen Vorschriften agieren und können folgende kritische Richtlinien abdecken:
- ISO 13485, FDA 21 CFR 820, FDA 21 CFR Part 11, SFDA etc.
- Zuverlässige und sichere (Herstellungs-) Prozesse
- Nachweis von Aktivitäten, um GxP-kritischen Prozessen zu entsprechen
Langfristig betrachtet hilft die Computer System Validation auch Kosten zu reduzieren:
- Geringer Wartungsaufwand durch Einsatz von Qualitäts- und Projektmanagementstandards
- Geringe Wartungsaufwände und Kosten im Change Management durch Transparenz und eine zuverlässige Systemdokumentation
- Geringe Kosten in der Entwicklung und Herstellung von Produkten durch Einsatz zuverlässiger Software und Prozesse
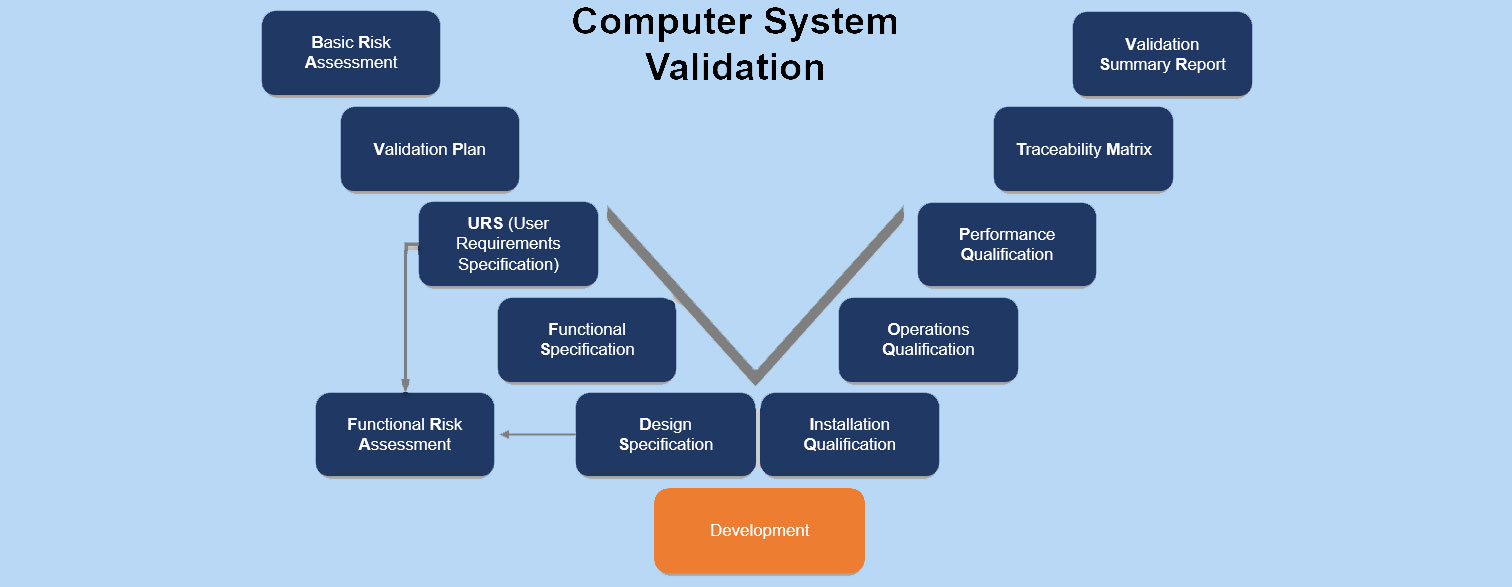
IQ, OQ und PQ im Kontext von Computer System Validation
Die Akronyme stehen für:
- IQ: Installation Qualification (Installationsqualifzierung)
- OQ: Operational Qualification (Funktionsqualifizierung)
- PQ: Performance Qualification (Leistungsqualifizierung).
Diese Qualifizierungen umfassen Aspekte der Validierung und der Qualifizierung der Software
-
IQ:
- Hardware Installation Qualification: Ist der dokumentierte Nachweis, dass
- die Anlage gemäß ihrer Spezifikation konzipiert und gebaut ist,
- die Anlage richtig installiert ist,
- die Anlage ihren festgelegten Zweck erfüllt,
- die Installation transparent ist,
- die Installation wiederholbar/reproduzierbar
- Software Installation Qualification:
- ist die Installationsanleitung/Richtlinie für das Computersystem samt unternehmensspezifischen Einstellungen
und Parametern - wird als Checkliste und Dokumentation für die richtige Installation verwendet,
- soll bei jeder Installation des Computersystems wiederverwendet werden (Clients, …),
- bietet eine Standardisierung der CS und ist geeignet für die wiederholte Verwendung einer Validierungsdokumentation in mehreren Systemen,
- jede Installation des CS ist identisch, transparent und wiederholbar/reproduzierbar.
- ist die Installationsanleitung/Richtlinie für das Computersystem samt unternehmensspezifischen Einstellungen
- Hardware Installation Qualification: Ist der dokumentierte Nachweis, dass
-
OQ:
- Die Betriebsqualifizierung (OQ) wird nach Erfüllung jedes Protokolls der IQ durchgeführt. Der Zweck der OQ ist es, festzustellen, dass die Leistung der Ausrüstung mit der Spezifikation der Benutzeranforderungen innerhalb der vom Hersteller angegebenen Betriebsbereiche übereinstimmt. In der Praxis bedeutet dies, dass Ausrüstungsmerkmale, die die Qualität des Endprodukts beeinflussen können, identifiziert und geprüft werden.
- Während der OQ werden alle Elemente des Prüfplans getestet und ihre Leistung wird gründlich dokumentiert. Da dies eine Voraussetzung für die Abnahme der Ausrüstung und der Anlage ist, kann sie erst durchgeführt werden, wenn die IQ gelaufen ist.
- Im Allgemeinen dient die OQ als detaillierte Überprüfung von Hardware- oder Software-Inbetriebnahme, Betrieb, Wartung, Reinigung und Sicherheitsprozeduren (falls und wo sie anwendbar sind). Für jede Einheit der Hard- und Software muss nachgewiesen werden, dass sie innerhalb der spezifizierten Grenzen arbeitet.
-
PQ:
- Der letzte Schritt der Qualifizierung von Geräten ist die PQ. In dieser Phase verifiziert und dokumentiert das Qualifizierungs- und Validierungsteam, dass die Ausrüstung mit reproduzierbaren Ergebnissen innerhalb eines bestimmten Arbeitsbereichs unter simulierten realen Bedingungen arbeitet. Anstatt die Komponenten und Geräte einzeln zu testen, werden sie in der PQ als Teil- oder Gesamtprozess geprüft.
- Bevor jedoch mit der Qualifizierung begonnen wird, muss das Team einen detaillierten Prüfplan auf der Grundlage der Prozessbeschreibung erstellen. Es ist wichtig zu wissen, dass die Qualität der Qualifizierung zu einem großen Teil von der Qualität des Testplans abhängt. Dies ist ein Bereich, in dem ein externer Spezialist hinzugezogen werden kann (und oft auch sollte), um Gründlichkeit und Genauigkeit sicherzustellen.
- Das Protokoll der Prozessleistungsqualifizierung (PPQ) ist ein grundlegender Bestandteil der Prozessvalidierung und -qualifizierung. Der Zweck ist es, eine kontinuierliche Produktqualität zu gewährleisten, indem die Leistung über einen bestimmten Zeitraum für bestimmte Prozesse dokumentiert wird.
Unser Leistungsspektrum
- Risiko basierende Validierung
- Validierung von SAP Softwarekomponenten und selbst entwickelten SAP Anwendungen
- Validierung gemäss ISO 13485
- Risikobewertung
- Testskripterstellung, Testausführung
- Durchführung der Validierung
- IQ/OQ/PQ
Computer System Validation gemäß V-Model
Risikomanagement in der Computer System Validation
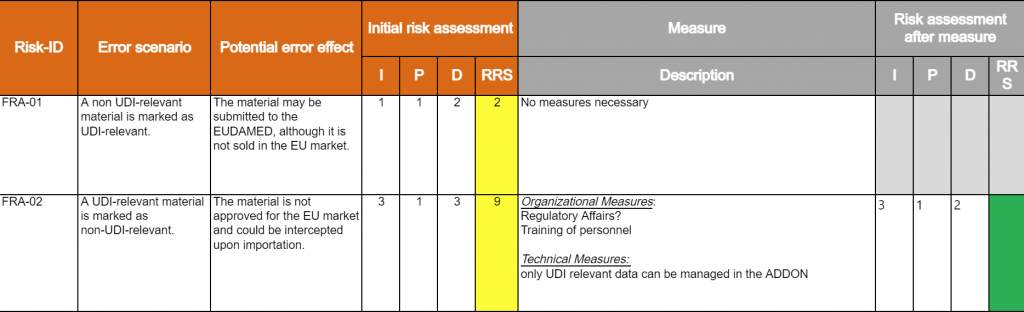
Das Risikomanagement ist eine wesentlicher Bestandteil des CSV und stellt die Qualität und Transparenz des Systems im Hinblick auf potentielle Risiken sicher. Im Rahmen der funktionale Risikobewertung (FRA) werden die Risiken im Bezug auf die Patientensicherheit, die Produktqualität und die Datenintegrität jeder Funktion bewertet.
- Bei dieser Risikobewertung werden unterschiedliche Risikoszenarien durchleuchtet und entsprechende Gegenmaßnahmen zur Reduzierung der Risiken definiert.
- Maßnahmen zur Risikominderung sollten vor allem dann definiert werden, wenn die Eintrittswahrscheinlich sowie der Einfluss des Risikos auf die Patientensicherheit, Produktqualität und Datenintegrität hoch sind und das Risiko im Use Case zudem nicht sofort erkannt werden kann.
- Basierend auf diesen Faktoren kann das potentielle Risiko berechnet und priorisiert werden.
Geeignete Gegenmaßnahmen zur Risikoreduzierung können auf zwei Ebenen definiert werden:
- Organisatorisch:
- Anwendung von Standardbetriebsprozesse (SOP)
- Trainings & Workshops für den sicheren Umgang mit der Software
- Technisch:
- Prüfung der Datenvalidität (Pflichtfelder, Business Rules)
- Prüfung der Datenintegrität (Cross-Checking)
Wenn Sie mehr über die Computer System Validation erfahren möchten, kontaktieren Sie uns gerne für einen individuellen Termin.
Nehmen Sie Kontakt mit uns auf
SAP Add-On mit Plugins für EUDAMED, FDA, SFDA, TGA, NMPA und weiteren Behörden:
Unser validiertes SAP UDI Add-On mit den Plugins erfüllt alle Anforderungen der weltweiten Behörden und repräsentiert eine effektive Lösung zur UDI Implementierung.