What is MDR/IVDR
Legislation
The new European Medical Device Regulation (MDR 2017/745) and the In Vitro Diagnostic Regulation (IVDR 2017/ 746) replace the existing medical device directives.

Since 25.05.2017, the EU regulations, the MDR and the IVDR, have come into force. After a postponement due to the COVID-19 pandemic, the MDR is now applicable from 26. 05.2021. The applicability of the IVDR was left as planned on 26.05.2022.
Aim of the MDR / IVDR
The aim of the new EU regulations is to further improve and increase patient safety. Accordingly, the main changes in MDR are as follows:
- Increased requirements for the qualification of test centre personnel, complete traceability of medical devices with the aid of clear labelling
- Increased requirements for the proof of clinical efficacy of products and the registration of all products in the central European database for medical devices (EUDAMED).
- Tightening of the requirements for market surveillance.
Timeline
The regulations on transitional provisions and transitional periods are formulated in a very complex manner. In addition, the postponement of EUDAMED by 2 years to 26.05.2022 and the above-mentioned postponement of the applicability of the MDR have repeatedly led to changes.
In the following section we use a graphic to show you when which legislation comes into force or must be implemented.
The date of registration of products depends on various factors. On the one hand, the classification of the products has an influence. In addition, a distinction is made as to whether the product is a directly marked product or whether the packaging is labelled.
For reusable products that are directly marked, the regulation grants two additional years. You can see the exact timetable in the graphically displayed schedule.
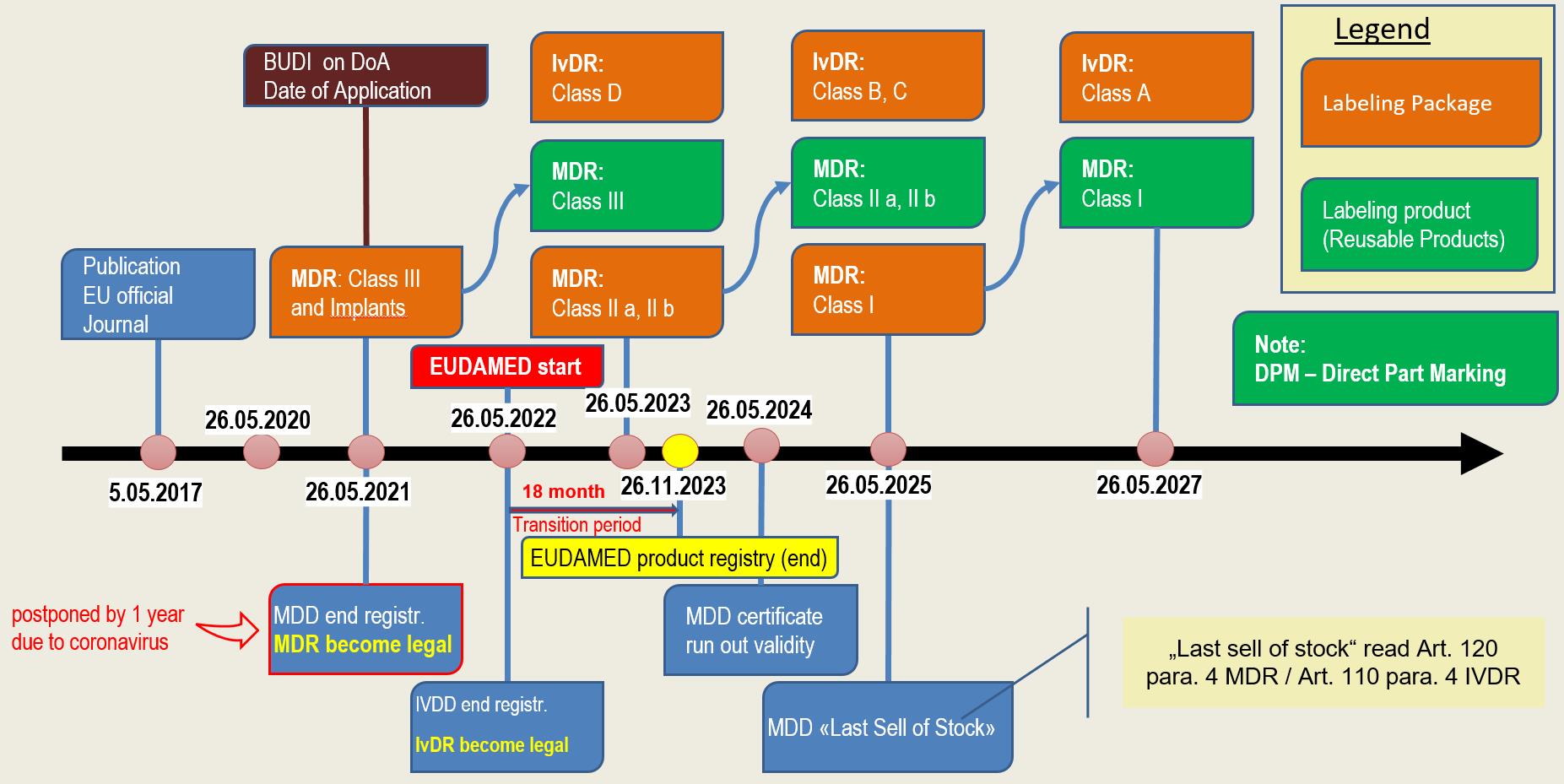
MDR & IvDR timeline (Updated version of 23.04.2020)
Changes
The effects of MDR are far-reaching. Various areas are affected, which presents the medical device industry with great challenges. The most important changes are:
Extension of the scope
The definitions in the field of medical devices and active implantable medical devices are considerably extended to include, for example, non-medical devices (e.g. coloured contact lenses, implants and materials for aesthetic purposes, etc.).
Designation of a “qualified person
Medical device manufacturers must appoint at least one person in their company who is ultimately responsible for ensuring that the requirements of the MDR are met. The company must be able to prove that this person has the necessary qualifications for the required tasks.
Implementation of the unique device identification system
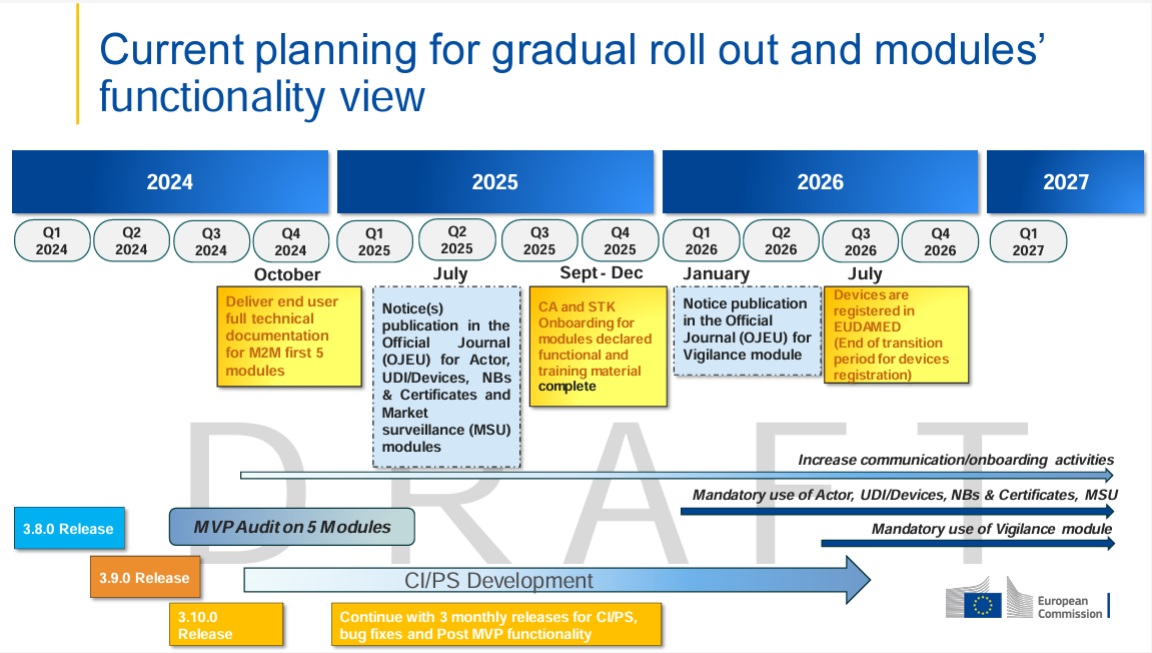
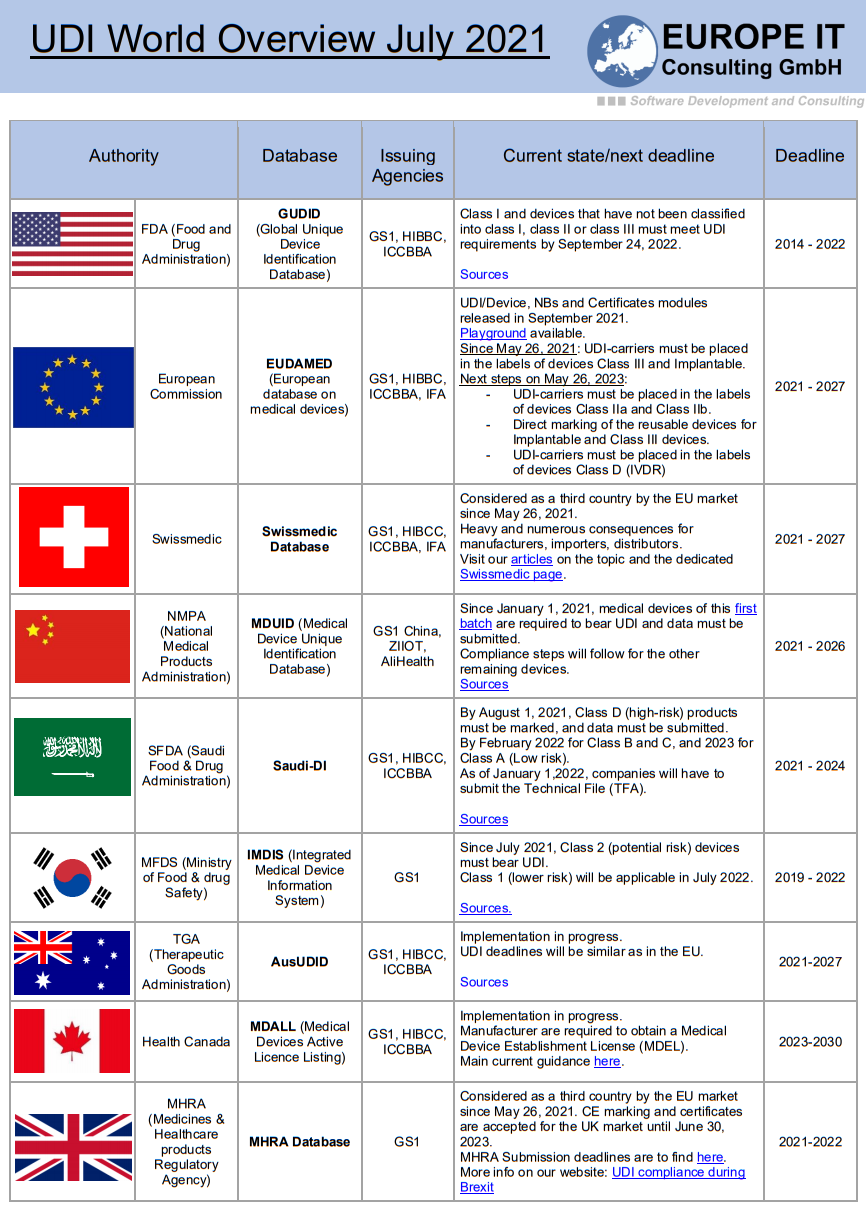
The legislation requires a system of unique product identification which is to be implemented with the UDI (Unique Device Identification) system. This should simplify the traceability of certain products in the supply chain for manufacturers and authorities. This will result in a faster and more efficient recall of medical devices that pose a safety risk. In this context, attention should also be paid to the labelling of products. This must comply with the conformity requirements of the MDR.
In addition, the European database for medical devices (EUDAMED) should be expanded and made more user-friendly. This should ensure easy access to information on approved medical devices. In order to register products, a Single Registration Number (SRN) must be applied from a notified body (Competence Authority, CA). Furthermore, manufacturers are obliged to provide the data of their products in digital form and to manage them. Later on, these must be transferred to the EUDAMED database.
Stricter clinical surveillance
With the powers of the new Regulation, Notified Bodies have further competences in post-market clinical surveillance. For example, unannounced audits, spot checks and product tests may now be carried out, which should help to reduce the risk posed by unsafe medical devices. For defined product groups, annual safety and performance reports must be submitted by manufacturers.
Re-classification of products
According to the new regulation, manufacturers must check the newly defined classification rules and adapt their technical documentation accordingly. Adjustments to the classification rules were mainly made in the areas of risk, contact duration and invasiveness. It should also be taken into account that even stricter clinical requirements apply to Class III medical devices and implantable devices. Regular monitoring is mandatory here.
The review of the classification and the adaptation of the technical documentation also includes the division into Basic UDI-DI and UDI-DI, which is required by the MDR. The BASIC UDI-DI is used to map all common characteristics of a product group. In contrast, the UDI-DI contains the product-specific information. There can be several UDI-DIs for one BASIC UDI-DI. Conversely, a UDI-DI is assigned to exactly one BASIC UDI-DI.
Stricter clinical evidence in the area of class III devices and implantable devices
As mentioned above, manufacturers who do not have sufficient clinical evidence to demonstrate the safety and performance of their device or appliance as required by the MDR must conduct full clinical investigations. There is also an obligation to collect and retain clinical data on potential safety risks through ongoing evaluations.
Re-clinical evaluation of Class IIa and IIb medical devices
Medical device manufacturers must systematically redo their clinical evaluations. In doing so, they must take into account the new formulations in the regulation on the equivalence of products. The circumstances under which a clinical trial could justifiably be dispensed with must also be reviewed.
No protection of the status quo
The legislation requires that all currently approved medical devices be re-certified according to the new requirements. Whether there will be any exceptions and which ones will apply is currently still under negotiation.
Impact
Considering the above mentioned points, medical device manufacturers are well advised to inform themselves about the legislative process of MDR at an early stage. They should regularly inform themselves about the additional changes that could affect them and initiate the implementation of these as soon as possible.
Due to the large number of medical devices that require testing and approval by a Notified Body, delays are to be expected. Do not lose any time and contact your Notified Body as soon as possible. In this way, a solution can be designed at an early stage to clarify any problems regarding conformity with the new legislation. Good preparation, careful planning and early initiation of measures are key elements for a smooth adaptation to the new requirements of the ordinance.







Related Posts