EUDAMED Fragen und Antworten
Inhaltsverzeichnis
Akteure & Rollen in EUDAMED
Produktregistrierung – Grundsätzliches & Fristen
- Fristen: Wann wird EUDAMED verpflichtend?
- Registrierungspflicht für Klasse-I-Produkte
- UDI-Registrierungspflicht für IVD-Produkte
- Registrierungspflicht für IVDD-Bestandsprodukte
Produktregistrierung – Praxis & Durchführung
- Einzelprodukt registrieren – Schritt-für-Schritt
- So funktioniert die Produkteintragung in EUDAMED
- Bereits registrierte Produkte bearbeiten
- Änderungen am Produkt (Name, Handelsname etc.)
- Warnhinweise & Kontraindikationen
- Basic UDI-DI nachträglich ändern
Datenstruktur & Pflichtangaben
- Gerätemodell – was eingeben?
- Pflichtdaten für die Produktregistrierung
- Marktinformationen – Datumsfelder
- Länder-Liste: Gilt nur für EU?
- „Ursprünglich in Verkehr gebracht“ – Bedeutung
Gruppierung & Kodierung
- Produkte unter einer Basic UDI-DI gruppieren
- GS1-Codes für Legacy Devices
- Fehlender EMDN-Code – was tun?
GUI & Systemfunktionen
- Funktionen in der EUDAMED-GUI
- Excel-Upload & Vermeidung doppelter Datenpflege
- Tool-Empfehlung bei Herstellerrolle
Nationale Systeme vs. EUDAMED
- Wird EUDAMED nationale Datenbanken ersetzen?
- Lokale Datenbanken, die weiterhin gepflegt werden müssen
Frage: Kann sich ein Unternehmen, das in EUDAMED als Importeur registriert ist, mit einem in EUDAMED registrierten Hersteller verknüpfen, ohne dass eine Benachrichtigung an den Hersteller gesendet und eine Genehmigung von ihm eingeholt wird?
Nein, das ist nicht ohne Wissen und aktive Mitwirkung des Herstellers möglich.
In EUDAMED ist die Verknüpfung von Akteuren – z. B. Importeur mit Hersteller – immer ein gegenseitiger Prozess, der eine Bestätigung bzw. Genehmigung durch den jeweils anderen Partner erfordert.Im Detail:
Der Importeur kann die Verknüpfung initiieren, indem er in seinem EUDAMED-Konto (im Actor-Modul) angibt, dass er mit einem bestimmten Hersteller verbunden ist (unter Angabe von dessen SRN).
Der Hersteller muss diese Verknüpfungsanfrage dann explizit annehmen. Ohne die Genehmigung durch den Hersteller wird die Verknüpfung nicht aktiv.
Frage: Die Mehrheit unserer Produkte fällt unter die Herstellerrolle, aber es gibt einige Produkte, die wir mit der Importeurrolle auf den Markt bringen. Für die Importeurrolle möchte ich wissen, wie die Registrierung erfolgen wird.
Registrierung als Importeur in EUDAMED – Schritt für Schritt
1. Importeurrolle verstehen (gemäß MDR, Artikel 13):
Ein Importeur ist ein Unternehmen mit Sitz innerhalb der EU, das ein Produkt von außerhalb der EU auf den EU-Markt bringt – also z. B. Produkte eines Nicht-EU-Herstellers zuerst in die EU einführt und vertreibt.
Wichtig:
Nur notwendig, wenn Ihr Produkte von einem Nicht-EU-Hersteller importiert.
Wenn Sie selbst Hersteller mit Sitz in der EU sind und eigene Produkte vertreibt, sind Sie kein Importeur im Sinne der MDR.
📝 Wie registriert man sich als Importeur?
A. Schritt 1: Registrierung im Actor-Modul
Gehen Sie auf das EUDAMED Actor Registration Portal
Wählen Sie „Importeur“ als Akteursrolle.
Reichen Sie die Akteurregistrierung bei eurer nationalen Behörde ein (in DE: BfArM, in FR: ANSM usw.).
Nach Genehmigung erhalten Sie Ihre SRN (Single Registration Number) als Importeur.
📌 Falls Sie bereits als Hersteller registriert seid, müssen Sie zusätzlich eine zweite Registrierung als Importeur durchführen – mit separater Rolle, aber ggf. gleichem Unternehmen.
B. Schritt 2: Verknüpfung mit Nicht-EU-Herstellern
Sobald Sie als Importeur eine SRN haben, können Sie sich mit den jeweiligen Nicht-EU-Herstellern verknüpfen, deren Produkte Sie in die EU importieren.
Die Verknüpfung erfolgt gegenseitig – der Hersteller muss zustimmen.
Diese Verknüpfung ist Voraussetzung dafür, dass die betreffenden Produkte in EUDAMED richtig zugeordnet werden.
📌 Wichtige Pflichten in der Importeurrolle (MDR Artikel 13):
Aufgabe Beschreibung Sicherstellen der MDR-Konformität Importeur muss kontrollieren, ob Produkt CE-gekennzeichnet ist, Gebrauchsanweisung vorliegt etc. UDI-Konformität prüfen Verpackung muss UDI-Label korrekt tragen Produktrückverfolgbarkeit sicherstellen Importeur muss seine Lieferkette dokumentieren UDI/Device-Einträge in EUDAMED prüfen Importierte Produkte müssen ordnungsgemäß registriert sein Zusammenarbeit mit Behörden Importeur muss Rückrufaktionen, Vorkommnisse etc. melden können
🧠 Besonderheit: Kombination Hersteller & Importeur
Wenn Sie z. B. eigene Produkte herstellwn (Herstellerrolle) und zusätzlich Produkte eines Nicht-EU-Herstellers unter eigener SRN importieren, dann:
müssen Sie sich zweimal registrieren: einmal als Hersteller, einmal als Importeur
müssen Sie für die importierten Produkte die Importeurpflichten separat erfüllen
dürfen Sie nicht dieselben Produkte gleichzeitig als Hersteller und Importeur registrieren – es muss klar zugeordnet sein
📋 Zusätzlicher Hinweis:
Für die Produktregistrierung ist bei importierten Produkten der Nicht-EU-Hersteller zuständig (Device-Modul).
Der Importeur wird im System als Economic Operator mitverknüpft, nicht als primärer Registrierender.
Frage: Wie ist die klare Abgrenzungen der Rollen bei EUDAMED.
Klare Abgrenzung der Rollen in EUDAMED – wer macht was?
In EUDAMED gibt es 6 Akteursrollen, aber für die Produktregistrierung und Pflichten sind diese 4 die wichtigsten:
Rolle SRN erforderlich? Produktregistrierung möglich? Typische Aufgaben Hersteller (Manufacturer) ✅ Ja ✅ Ja Registriert Produkte (UDI), verwaltet Device-Daten, Vigilanz Bevollmächtigter (Authorised Representative) ✅ Ja Nein (außer wenn vom Hersteller delegiert) Vertritt Nicht-EU-Hersteller, kann administrative Aufgaben übernehmen Importeur (Importer) ✅ Ja ❌ Nein Verknüpft sich mit Hersteller, übernimmt MDR-Pflichten (z. B. Etikettenprüfung, Rückverfolgbarkeit) System-/Prozedurpackungs-Hersteller ✅ Ja ✅ Ja Meldet Sets, die aus mehreren Produkten bestehen 🔍 1. Die Rolle „Hersteller“ (Manufacturer)
Hat die volle Kontrolle über die Produktregistrierung
Muss jede Basic UDI-DI und UDI-DI registrieren
Kann sowohl EU- als auch Nicht-EU-Hersteller sein (Nicht-EU mit Mandat durch EU-Bevollmächtigten)
Verknüpft Produkte mit Economic Operators (Importeure, Distributoren)
Verantwortlich für Vigilanzmeldungen, techn. Dokumentation, Zweckbestimmung etc.
🔍 2. Die Rolle „Importeur“
Kann sich nur registrieren, wenn der Firmensitz innerhalb der EU liegt
Registriert keine Produkte selbst – aber muss:
Sich mit dem Hersteller verknüpfen
Sicherstellen, dass die Produkte, die er einführt, in EUDAMED registriert sind
Seine eigene Firma im Economic Operator-Modul sichtbar machen
Importeur muss seine SRN auf dem Etikett anbringen (Art. 13 MDR)
📌 Ein und dieselbe Firma kann sowohl Hersteller als auch Importeur sein – aber in EUDAMED sind das zwei getrennte Rollen mit jeweils eigener SRN.
🔍 3. Die Rolle „Bevollmächtigter“ (Authorised Representative)
Notwendig, wenn der Hersteller nicht in der EU ansässig ist
Wird durch eine Mandatserklärung mit dem Nicht-EU-Hersteller verbunden
Registriert keine Produkte selbst, außer er übernimmt diese Aufgabe im Namen des Herstellers
👥 Verknüpfung der Rollen – Beispiel aus der Praxis
Wenn ihr also:
Hersteller seid mit Sitz in der EU → Ihr registriert die Produkte selbst (UDI-Daten etc.)
Gleichzeitig Importeur für Produkte eines US-Herstellers → Ihr registriert euch separat als Importeur, bekommt eigene SRN und verknüpft euch mit dem US-Hersteller
📄 Kurz und knapp: Was bedeutet das für euch?
Wenn ihr… Dann müsst ihr… eigene Produkte in der EU herstellt Euch als Hersteller registrieren, Produkte selbst eintragen Produkte aus Drittstaaten in die EU importiert Euch zusätzlich als Importeur registrieren und mit dem Hersteller verknüpfen für Nicht-EU-Hersteller agiert Entweder als Bevollmächtigter auftreten oder eine vertragliche Verbindung sicherstellen
🎯 Fazit:
EUDAMED ist rollenscharf aufgebaut – und jede Rolle hat eigene Aufgaben, Pflichten und eine eigene SRN. Sobald ein Unternehmen mehrere Rollen einnimmt, braucht es klare interne Prozesse und Systemzuordnung, um Verwechslungen oder Audit-Probleme zu vermeiden.
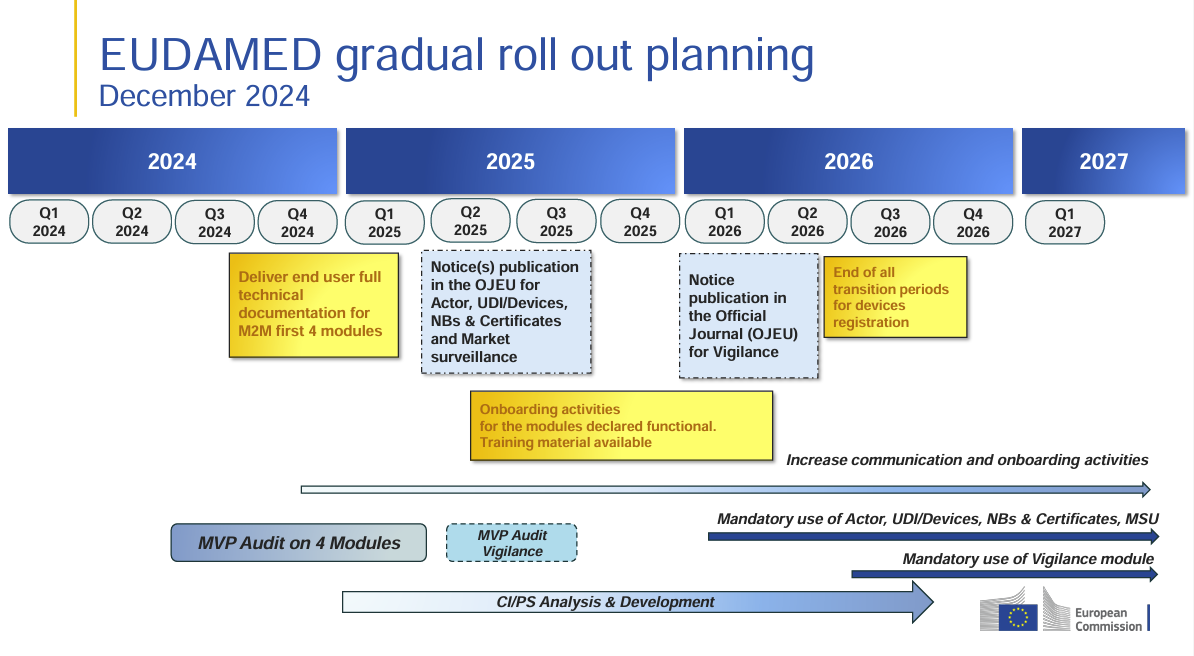
Frage: Fristen: Die EU-Kommission hat angekündigt, dass das UDI-Registrierungsmodul bald vollständig funktionsfähig sein wird, wobei die Frist für die Registrierung der Produkte Anfang nächsten Jahres liegt. Wird erwartet, dass diese Frist eingehalten wird und EUDAMED rechtzeitig einsatzbereit ist?
Stand: Q1/2025:
Ja, es wird erwartet, dass die EU-Kommission den vollständigen Betrieb des EUDAMED-UDI/Device-Moduls in der ersten Jahreshälfte 2025 im Amtsblatt der EU (OJEU) offiziell bestätigt. Die Frist zur verpflichtenden Produktregistrierung wird dann voraussichtlich 24 Monate nach dieser Bekanntmachung liegen.
👉 Das bedeutet: Die Frist wird frühestens Anfang 2027 enden.
📅 Was ist aktuell der Stand (März 2025)?
Das UDI/Device-Modul ist technisch bereits voll funktionsfähig und freiwillig nutzbar.
Die EU-Kommission führt aktuell die letzten Interoperabilitätstests (M2M) mit Wirtschaftsteilnehmern und nationalen Behörden durch.
Die offizielle Bekanntmachung im EU-Amtsblatt (OJEU) steht noch aus – sie markiert den „Startschuss“ für die 24-monatige Übergangsfrist.
🧠 Was bedeutet das für Hersteller & Importeure?
Thema Bedeutung Freiwillige Nutzung Jetzt schon möglich & empfohlen – hilft bei interner Prozesssicherheit und Datenpflege Verpflichtende Nutzung Startet erst nach OJEU-Bekanntmachung → aktuell keine rechtliche Pflicht UDI-Registrierungspflicht 24 Monate nach OJEU-Veröffentlichung für alle MDR-Produkte erforderlich Empfehlung Jetzt bereits mit der Registrierung beginnen – um Engpässe und Fehler zu vermeiden 📌 Praxistipp:
Viele Unternehmen unterschätzen den Aufwand für die UDI-Datenpflege, v. a. bei großen Portfolios. Wer bis zur Verpflichtung wartet, läuft Gefahr, den Aufwand in der Übergangsfrist nicht zu schaffen – oder in Audits Probleme zu bekommen.
💬 Fazit:
Die Frist wird voraussichtlich im Amtsblatt veröffentlicht – vermutlich im Laufe von 2025.
Die Registrierungspflicht gilt ab dann + 24 Monate, also vermutlich ab Anfang 2027.
Technisch ist EUDAMED bereit – Unternehmen sollten sich jetzt vorbereiten, selbst wenn die Pflicht noch nicht greift.
Frage: Wie lauten die Fristen für die Registrierung von Klasse I Produkten?
Die Registrierungspflicht für Klasse I Produkte in EUDAMED beginnt erst, wenn die EU-Kommission die vollständige Funktionsfähigkeit von EUDAMED offiziell im Amtsblatt (OJEU) veröffentlicht. Ab diesem Zeitpunkt gilt eine Übergangsfrist von 6 Monaten.
📅 Das bedeutet konkret:
Schritt Datum OJEU-Veröffentlichung (vollständige EUDAMED-Funktion) ⚠️ Noch ausstehend (voraussichtlich 2025) Start der Pflicht zur Produktregistrierung (Klasse I) 📆 6 Monate nach OJEU-Veröffentlichung Registrierung muss abgeschlossen sein 📆 Voraussichtlich Anfang/Mitte 2026, je nach Veröffentlichungsdatum
Frage: muss ich die EUDAMED für die UDI-Kennzeichnung (nach IVDR) bereits nutzen bzw. ab wann ist es notwendig?
Nein, aktuell besteht noch keine rechtliche Verpflichtung, UDI-Daten in EUDAMED zu registrieren – auch nicht unter IVDR.
ABER:
Die EU-Kommission wird voraussichtlich im Laufe von 2025 die vollständige Funktionsfähigkeit von EUDAMED (einschließlich UDI-Modul) im Amtsblatt der EU bekanntgeben (OJEU).
👉 Ab diesem Zeitpunkt läuft eine 24-monatige Übergangsfrist.Das bedeutet:
Die UDI-Registrierung in EUDAMED wird ab frühestens 2027 verpflichtend – auch für IVD-Produkte.
Frage: Sind auch Bestandsprodukte nach IVDD dort zu hinterlegen?
Nein, Bestandsprodukte, die noch unter der IVDD in Verkehr gebracht wurden, müssen aktuell nicht in EUDAMED registriert werden.
Die UDI-Registrierungspflicht gilt nur für IVDR-konforme Produkte.Übergangsfrist & Ausnahmen
🧾 Was sind „Bestandsprodukte“?
Das sind Produkte, die nach IVDD zertifiziert wurden und gemäß den Übergangsfristen der IVDR Artikel 110 weiterhin in Verkehr gebracht werden dürfen – z. B. auf Grundlage einer alten Notified-Body-Bescheinigung. Solche Produkte dürfen je nach Klasse bis spätestens Mai 2026 bzw. Mai 2027 weiter vermarktet werden, ohne dass sie bereits IVDR-konform sein müssen.
❗ Für diese Produkte gilt:
Sie benötigen noch keine UDI-Kennzeichnung
Sie müssen nicht in EUDAMED registriert werden
Es entstehen keine neuen Pflichten in EUDAMED – weder für das Device-Modul noch für die Vigilanz
🚦 Wann müssen IVD-Produkte in EUDAMED registriert werden?
Produkttyp Registrierung in EUDAMED erforderlich? Neues IVDR-konformes Produkt ✅ Ja, nach Start der UDI-Pflicht (ab 2027 erwartet) Bestandsprodukt nach IVDD ❌ Nein, solange es nach Übergangsregel weiterverkauft wird Umgestelltes Produkt (IVDR-Neuzertifizierung) ✅ Ja
Frage: Wie registriert man ein einzelnes Produkt der Klasse I?
Einzelprodukt in EUDAMED registrieren – so geht’s:
Einloggen:
https://webgate.ec.europa.eu/eudamed/UDI/Devices-Modul öffnen
„Register new UDI/Device“ wählen
Schrittweise durch den Wizard gehen (5 Schritte):
Schritt 1: Basic UDI-DI, Device Name, EMDN, Risikoklasse, Zweckbestimmung
Schritt 2: UDI-DI (GTIN), Verpackung, Sterilität
Schritt 3: Economic Operators (Hersteller, ggf. Bevollmächtigter/Importeur)
Schritt 4: Marktverfügbarkeit („von“-Datum, ggf. „bis“-Datum)
Schritt 5: Validieren & abschicken (Submit)
Status prüfen: Produkt wird als „Submitted“ oder „Published“ angezeigt
Frage: Wie genau macht man Produkteintragungen in die EUDAMED?
Voraussetzung:
Frage: Wie können registrierte Produkte nachträglich bearbeitet werden?
✅ So bearbeiten Sie ein bereits registriertes Produkt in EUDAMED:
Einloggen in EUDAMED
👉 https://webgate.ec.europa.eu/eudamedUDI/Devices-Modul öffnen
Produkt über UDI-DI oder Basic UDI-DI suchen
Rechts neben dem Eintrag auf „Actions“ → „Update“ klicken
Den Assistenten (Wizard) durchgehen und die gewünschten Felder ändern
Validieren und dann Submit – das Update wird eingereicht und verarbeitet
📌 Wichtig:
Sie können nur Produkte bearbeiten, die mit deiner SRN als Hersteller verknüpft sind
Manche Felder (z. B. Basic UDI-DI) sind nicht änderbar – dafür ggf. neues Produkt anlegen
Frage: Die Zusammen zwischen den Aufgaben (z.B. Ändern des Produktnamen oder Hinzufügen eines Handelsname) und die damit verbundenen Tätigkeiten Aktionen mit dem GUI der EUDAMED sowie die sinnvolle Reihenfolge der Tätigkeiten/Aktionen im GUI.
Typische Änderungsbeispiele im GUI
Änderung Betroffenes Feld (GUI) EUDAMED-Modul Erforderliche Aktion Produktname ändern „Device Name“ UDI/Device Modul Update Submission Handelsname hinzufügen „Brand Name“ UDI/Device Modul Update Submission Modellnummer anpassen „Model“ UDI/Device Modul Update Submission Zweckbestimmung (Intended Purpose) anpassen „Intended Use“ UDI/Device Modul Update Submission Verpackungsinformationen ergänzen „Packaging“ UDI/Device Modul Update Submission Sterilität/Label-Daten ändern „Device Characteristics“ UDI/Device Modul Update Submission 🖥️ Ablauf im GUI – empfohlene Reihenfolge der Schritte
🎯 Szenario: Sie möchten Produktname UND Handelsname ändern/hinzufügen
🔄 1. Einloggen & Produkt auswählen
Gehen Sie auf das UDI/Device-Modul
Suchen Sie nach der betreffenden UDI-DI (GTIN)
Klicken Sie auf „Actions“ → „Update“
✍️ 2. Schrittweise Anpassung im Wizard
Die GUI ist in Schritte unterteilt – folgende Felder sind relevant:
Schritt 1: Basic UDI-DI & Device Information
Hier können Sie Device Name (Produktname) und Brand Name (Handelsname) anpassen
Auch können Sie hier mehrere Handelsnamen hinzufügen (z. B. bei multilingualen Märkten)
Schritt 2: Device Characteristics
Prüfen Sie, ob durch die Namensänderung weitere Daten betroffen sind (z. B. Etikettensprache, Verpackung)
Schritt 3–5: Review und Submit
Nach der letzten Seite können Sie Ihre Eingaben überprüfen
Klicken Sie auf „Validate“ → das System prüft, ob alle Pflichtfelder korrekt sind
- Danach auf „Submit“
🔐 3. Nach dem Submit
- Der Status der Änderung wird als „Under Review“ oder „Updated“ angezeigt
- Manche Änderungen erscheinen sofort, andere erst nach Freigabe durch die zuständige Behörde (je nach Feld und nationaler Umsetzung)
⚠️ Wichtige Hinweise zur Reihenfolge & Logik
- Device Name vs. Brand Name:
- Device Name = technische Produktbezeichnung
- Brand Name = kommerzielle Marke auf dem Markt (z. B. „MediPlus®“)
- Brand Name kann mehrfach sein, Device Name nicht → Brand Name also mit Bedacht wählen
- Änderungen an Produktname oder Brand Name können Rückwirkungen auf Label, IFU oder techn. Doku haben – also vorher intern abstimmen
- Sie können mehrere Änderungen in einem Update Submission zusammenfassen – das ist effizienter, als alles einzeln einzureichen
Frage: Könnten Sie bitte Beispiele für kritische Warnhinweise oder Kontraindikationen (Schritt 4: UDI-DI-Merkmale) geben?
In diesem Feld soll der Hersteller relevante Warnungen oder Kontraindikationen zum Produkt angeben, sofern vorhanden. Das Ziel ist es, Patienten- und Anwendersicherheit direkt sichtbar zu machen.
✅ Beispiele für typische kritische Warnhinweise:
„Nicht für die Anwendung bei Patienten mit implantiertem Defibrillator geeignet.“
„Nicht in der Nähe von Magnetresonanztomografen (MRT) verwenden.“
„Nur durch geschultes Fachpersonal anwenden.“
„Steril – bei beschädigter Verpackung nicht verwenden.“
✅ Beispiele für Kontraindikationen:
„Nicht anwenden bei bekannter Allergie gegen Latex.“
„Nicht geeignet für Kinder unter 3 Jahren.“
„Nicht verwenden bei offenen Wunden oder Hauterkrankungen im Applikationsbereich.“
„Nicht verwenden bei schwangeren oder stillenden Frauen.“
⚠️ Wichtig:
Wenn keine Warnhinweise oder Kontraindikationen vorliegen, kann das Feld mit „Keine bekannt“ ausgefüllt werden – aber nie leer lassen.
Die Eingabe erfolgt als Freitext, also solltest du präzise, aber knapp formulieren.
Frage: Ich möchte wissen, wie man eine Basic UDI-DI in EUDAMED am besten korrigieren oder ändern kann. Ich habe versucht, einen neuen Eintrag mit einer anderen Basic UDI-DI für ein zuvor registriertes Produkt zu erstellen. Allerdings erhalte ich eine Fehlermeldung (Schritt 3: UDI-DI-Identifikationsinformationen), da der UDI-DI-Code/GTIN des Geräts bereits für meinen vorherigen Eintrag verwendet wurde.
Die Basic UDI-DI ist das übergeordnete Identifikationsmerkmal einer Produktgruppe (z. B. gleiche Zweckbestimmung, Risikoklasse, essentielles Design).
Sie ist fest mit dem Produkt-UDI-DI (GTIN) verknüpft und kann nach Einreichung nicht einfach ausgetauscht werden, wenn bereits ein Eintrag mit dieser GTIN existiert.🔒 Weshalb erscheint die Fehlermeldung?
Wenn Sie versuchen, ein Produkt mit einer neuen Basic UDI-DI zu registrieren, aber dabei eine UDI-DI (GTIN) verwenden, die bereits mit einer anderen Basic UDI-DI eingereicht wurde, erkennt EUDAMED das als Inkonsistenz und blockiert den Vorgang:
Fehler: Der UDI-DI-Code wurde bereits für eine andere Basic UDI-DI verwendet.
Lösungsmöglichkeiten:
1. Bestehenden Eintrag aktualisieren, nicht neu einreichen
Wenn die UDI-DI/GTIN bereits registriert ist, können Sie nur über eine Änderung des bestehenden Eintrags gehen – z. B. durch eine Update Submission (über Webinterface oder M2M/XML), nicht durch eine neue Registrierung.
👉 Aber Achtung: Die Basic UDI-DI selbst kann bei einem bestehenden Gerät nicht geändert werden.
2. Wenn die Basic UDI-DI wirklich falsch war (z. B. Tippfehler)
Dann gibt es nur zwei Möglichkeiten:
Gerät abmelden (Deactivate/Delete) und neu registrieren mit korrekter Basic UDI-DI und UDI-DI
Oder (falls das Gerät noch nicht aktiv vermarktet wurde): eine neue UDI-DI generieren (neue GTIN) und mit der korrekten Basic UDI-DI neu registrieren
📌 Wichtig: Die Verknüpfung UDI-DI ↔ Basic UDI-DI ist dauerhaft. Es kann nicht dieselbe GTIN mit zwei unterschiedlichen Basic UDI-DIs existieren.
3. Wenn mehrere ähnliche Produkte registriert werden sollen:
Dann sollten sie jeweils eine eigene GTIN erhalten – auch wenn sie sich in Details unterscheiden – und über eigene Basic UDI-DIs registriert werden, sofern gerechtfertigt (unterschiedliche Zweckbestimmung, Risikoklasse etc.).
Frage: Könnten Sie bitte beschreiben, welche korrekten Informationen im Feld ‚Gerätemodell‘ (Schritt 1: Basic UDI-DI-Informationen) eingegeben werden müssen? Ich habe festgestellt, dass Hersteller dieses Feld in EUDAMED nicht einheitlich verwenden.
Definition laut MDCG 2018-1:
Das Gerätemodell („Device Model“) beschreibt die produkttechnische oder kommerzielle Bezeichnung, unter der das Medizinprodukt identifizierbar vermarktet oder verwendet wird. Es hilft, Produkte innerhalb einer Basic UDI-DI-Gruppe zu unterscheiden.✅ Richtig wäre also:
Eine technische Modellbezeichnung, die im Etikett, in der Gebrauchsanweisung oder Produktdokumentation genannt wird.
Oder eine Artikelnummer-bezogene Bezeichnung, wenn keine Modellnummer vorhanden ist.
❌ Nicht geeignet sind:
Produktbeschreibungen („Steriles Einweg-Katheterset für Kinder“)
Zweckbestimmungen („Zur Behandlung von XYZ“)
Freitexte mit Schlagwörtern oder Variantenkombinationen
🔍 Beispiele für korrekte Gerätemodell-Einträge:
Produkttyp Gerätemodell Herzschrittmacher Model ACURA 3000 Einweg-OP-Maske Type 2R – MaskCare Blue Dentalimplantat IMPL-24-Titanium Software (SaMD) Diagware v1.4.2 Blutdruckmessgerät BP-Monitor Pro M120
Frage: Welche Datenelemente für die Produktregistrierung sind zwingend?
Pflichtfelder bei der Produktregistrierung in EUDAMED (UDI-DI-Ebene)
Hier nach Kategorien geordnet:📘 A. Basic Device Information (Schritt 1 im GUI)
Feld Pflicht? Kommentar Basic UDI-DI ✅ Ja Vom Hersteller selbst vergeben (z. B. über GS1) Device Name ✅ Ja Technischer Name (kein Marketingtext) Brand Name ❌ Nein Nur wenn vom Markt verwendet EMDN Code ✅ Ja EU-Nomenklaturcode (mind. 4-stellig) Regulatory Status (MDR/IVDR) ✅ Ja Gibt an, ob MDR oder IVDR Risk Class ✅ Ja I, IIa, IIb, III (MDR) oder A–D (IVDR) Intended Purpose ✅ Ja Zweckbestimmung in Klartext Notified Body Information ✅ Ja, wenn zutreffend Nur bei Produkten mit NB-Zertifikat 📦 B. UDI-DI & Packaging (Schritt 2 im GUI)
Feld Pflicht? Kommentar UDI-DI (GTIN o. ä.) ✅ Ja Hauptidentifikator auf der Verpackung Primary Packaging Type ✅ Ja Z. B. Einzelverpackung, Bulk etc. Sterility Information ✅ Wenn Produkt steril ist Sonst kann leer bleiben Storage & Shelf Life ❌ Optional Nur bei Temperatur-/Verfallsabhängigkeit Critical Warnings / Contraindications ❌ Optional Falls vorhanden, eintragen 👥 C. Economic Operators (Schritt 3 im GUI)
Feld Pflicht? Kommentar Hersteller (SRN) ✅ Ja Automatisch, wenn du selbst Hersteller bist Bevollmächtigter ✅ Wenn du nicht-EU-Hersteller bist Muss gültige SRN haben Importeur ❌ Optional Nur falls vorhanden – sonst leer lassen 🌍 D. Market Information (Schritt 4 im GUI)
Feld Pflicht? Kommentar „Von“-Datum (Date from) ✅ Ja Startdatum der Marktverfügbarkeit „Bis“-Datum (Date to) ❌ Nein Nur bei Abkündigung/Marktrücknahme 📎 E. Anhänge (Schritt 5 im GUI)
Feld Pflicht? Kommentar Label / IFU Upload ❌ Optional Kann bei Audits verlangt werden, aber nicht verpflichtend Technische Doku ❌ Nicht direkt in EUDAMED Nur bei Behördenanfrage erforderlich
Frage: Bitte um eine Klärung bezüglich des Abschnitts „Marktinformationen“ in EUDAMED, insbesondere in Bezug auf die Datumsfelder „von (JJJJ-MM-TT)“ und „bis (JJJJ-MM-TT)“. Wofür stehen diese Daten genau bzw. müssen diese ausgefüllt werden?
Das hängt von der Gesetzgebung ab, unter der Sie Ihr Produkt registrieren. Wenn Sie die Alt-Gesetzgebung (MDD, IVDD, AIMDD) auswählen, müssen Sie mindestens das „gültig bis“-Datum eingeben (optional können Sie auch das Startdatum angeben). Aber nur das „bis“-Datum ist verpflichtend. Wenn Sie hingegen die neue Verordnung wie „MDR“ oder „IVDR“ auswählen, müssen Sie gar kein Datum angeben.
Wofür stehen die Felder „von (YYYY-MM-DD)“ und „bis (YYYY-MM-DD)“ im Abschnitt „Market Information“?
Feld Bedeutung „von“-Datum ( Date from which the device is/was made available on the market)Das Datum, ab dem das Produkt erstmalig auf einem EU-Markt verfügbar gemacht wurde (gemäß Artikel 2 Nr. 27 MDR/IVDR: „Inverkehrbringen“) „bis“-Datum ( Date until which the device is/was made available on the market)Das Datum, bis zu dem das Produkt aktiv auf dem Markt ist bzw. war – z. B. bei Rückzug vom Markt, Produktauslauf, Deaktivierung 🧠 Was bedeutet das konkret in der Praxis?
Das „von“-Datum ist typischerweise das Launch-Datum in der EU
Das „bis“-Datum wird nur gesetzt, wenn das Produkt nicht mehr aktiv vermarktet wird oder ein festes Ablaufdatum hat
📌 Beispiel:
Ein Produkt wurde am 01.01.2023 auf den Markt gebracht und ist weiterhin verfügbar:
→ „von“ = 2023-01-01
→ „bis“ = leer lassen (nicht erforderlich)⚠️ Sind die Felder verpflichtend?
▶️ Aktueller Stand laut EU-Kommission (XSD v2.0 & GUI-Spezifikation):
Feld Pflichtfeld? „von“-Datum ✅ Ja, verpflichtend „bis“-Datum ❌ Nein, nur bei Abkündigung oder Marktende erforderlich
Frage: Abschnitt 2 „Liste aller Länder, in denen das Produkt verfügbar ist oder sein wird“, gilt das nur für EU-Länder?
Ja, in diesem Abschnitt sind auch nur EU-Länder auswählbar.
Frage: Angabe, dass ein Land als „ursprünglich in Verkehr gebracht“ definiert werden muss, was ist damir gemeint? Jenes Land, in dem das Medizinprodukt (UDI-Datensatz) erstmals auf den Markt gebracht wurde?
Bezieht sich diese Angabe ausschließlich auf ein erstes EU-Mitgliedsland, oder können in diesem Feld auch Drittstaaten außerhalb der EU berücksichtigt werden?
EUDAMED bezieht sich ausschließlich auf den EU-Markt. Länder außerhalb der EU sind nicht Teil dieses Systems. Hersteller, die ihre Produkte auch in Nicht-EU-Staaten vertreiben möchten, müssen die jeweiligen regulatorischen Anforderungen dieser Länder separat erfüllen.
Frage: Ist es ratsam, dass Produkte nach Basic UDI s zusammengeführt und registriert werden? (Clustern oder separat), z.B. Schläuche für Insufflatoren – Stichwort: Ist Gruppierung von Produkten zum Minimieren von Arbeitsaufwand möglich?
Ja, eine sinnvolle Gruppierung („Clustering“) von Produkten unter einer gemeinsamen Basic UDI-DI ist nicht nur zulässig, sondern auch ausdrücklich vorgesehen – solange bestimmte Kriterien erfüllt sind. Das reduziert deutlich den Registrierungs- und Pflegeaufwand in EUDAMED.
📘 Was ist die Basic UDI-DI und wofür dient sie?
Die Basic UDI-DI ist:
der Primärschlüssel für die Produktgruppe
die Verbindung zur Konformitätserklärung, Zertifizierung, techn. Dokumentation
nicht auf der Verpackung abgedruckt, sondern nur in EUDAMED & Doku
📦 Wann dürfen Produkte unter eine gemeinsame Basic UDI-DI gruppiert werden?
Laut MDCG 2018-1 (und EU-Kommission):
✅ Die Produkte müssen:
Kriterium Bedeutung Gleiche Zweckbestimmung Sie erfüllen denselben klinischen oder diagnostischen Zweck Gleiche Risikoklasse z. B. alle Klasse IIa Ähnliche Grundkonstruktion / Design z. B. Schläuche aus gleichem Material, gleichem Anschluss Teil derselben techn. Dokumentation Sie werden in einer Datei bewertet, z. B. zusammen in einer DoC 🧪 Beispiel: Schläuche für Insufflatoren
Diese können problemlos gruppiert werden, wenn:
sie dieselbe Funktion haben (z. B. Verbindung von Insufflator mit Patient)
sich nur in Länge, Farbe oder Verpackungseinheit unterscheiden
der gleiche Hersteller sie produziert und eine gemeinsame technische Doku vorliegt
👉 Dann ist es ratsam, diese Varianten unter einer Basic UDI-DI zu clustern und mehrere UDI-DI (GTINs) unter dieser Basis zu registrieren.
🎯 Vorteile des Clusterns (Gruppierung):
Vorteil Beschreibung 🔁 Weniger Aufwand Nur eine Registrierung auf Basis-Ebene mit mehreren UDI-DIs 🧾 Einheitliche Doku Nur eine Konformitätserklärung & technische Akte notwendig 🔍 Bessere Übersicht In EUDAMED kannst du deine Varianten strukturierter verwalten 📈 Skalierbarkeit Neue Varianten können später einfacher ergänzt werden ⚠️ Aber Achtung: Wann Sie nicht clustern sollten:
Nicht clustern bei… Grund Unterschiedliche Zweckbestimmung z. B. Schlauch für CO₂ vs. für andere Gase Unterschiedliche Risikoklassen z. B. Klasse I vs. Klasse IIa Unterschiedliche regulatorische Bewertungen z. B. eine Variante steril, die andere nicht Unterschiedliche Zertifikate oder Notified Bodies → getrennte Basic UDI-DIs erforderlich
Frage: Dürfen wir die Basic UDI-Dis von GS1 für Legacy Devices nutzen oder müssen wir die von Eudamed generieren lassen? Das bedeutet, die letzteren müssen mit den neuen verknüpft werden?
Ja, Sie dürfen GS1-generierte Basic UDI-DIs auch für Legacy Devices verwenden – eine automatische Generierung durch EUDAMED ist nicht notwendig.
👉 Sie müssen die Basic UDI-DI selbst vergeben (z. B. über GS1) – auch für Legacy-Produkte –, wenn Sie diese in EUDAMED registrieren wollen (freiwillig oder später verpflichtend).
Frage: Gibt es eine Möglichkeit neue EMDN Codes zu generieren, wenn sich ein neues Produkt mit einer neuen Technologie in den vorhandenen Codes nicht wiederfinden lässt?
Nein, Hersteller können keine eigenen EMDN-Codes generieren. Neue Codes können nur von der EU-Kommission hinzugefügt werden – auf Antrag bzw. über definierte Kanäle.
Was tun, wenn es keinen passenden EMDN-Code gibt?Den nächstgelegenen EMDN-Code wählen
Sie sind verpflichtet, den bestmöglich zutreffenden existierenden Code zu wählen
Wenn Ihr Produkt z. B. ein innovatives tragbares Dialysegerät ist, aber es gibt nur Codes für klassische Dialysemaschinen – dann nimm den am nächsten liegenden
In der Regel reichen 4-stellige oder 6-stellige Codes (Level 2 oder 3) für die Produktregistrierung aus
📌 Diese Auswahl muss gut dokumentiert werden – auch für Audits (z. B. warum du diesen Code gewählt hast).
Frage: Was kann man noch sonst mit der GUI machen, z.B. kann man nach bestimmten Produkten, Produktklassen, Herstellern, Importeuren, etc. suchen. Die Suche liefert oft leere Antworten.
Typische Funktionen im UDI/Device-Modul:
Funktion Beschreibung Bemerkung 🔍 Produkte suchen Nach UDI-DI (GTIN), Basic UDI-DI, Device Name, Brand Name Oft case-sensitive und sehr restriktiv 🗂️ Produktdetails anzeigen Alle Details zum registrierten Produkt inkl. Verpackung, Label, Intended Use etc. Nur bei korrektem Suchtreffer ✏️ Produkt ändern (Update Submission) Produktinformationen ändern oder ergänzen Nur für Produkte, die von der eigenen SRN stammen 🗑️ Produkt deaktivieren (Deactivate) Produkt vom Markt nehmen oder zurückziehen Nur durch den registrierenden Hersteller möglich ⬆️ Neues Produkt registrieren Mit vollständigem Wizard über 5 Schritte Basic UDI-DI muss korrekt sein ⏳ Produktstatus prüfen „Submitted“, „Under Review“, „Published“ etc. Wichtiger Überblick über Workflow-Status Typische Funktionen im Actor-Modul:
Funktion Beschreibung Bemerkung 🔍 Akteure suchen Nach SRN, Unternehmensname, Rolle (Hersteller, Importeur etc.) Funktioniert oft nur mit exakter Schreibweise 🔗 Verknüpfung mit anderen Akteuren z. B. Hersteller mit Importeur oder Bevollmächtigtem Gegenseitige Bestätigung erforderlich 🧾 Actor-Details anzeigen Adresse, Rollen, Zuständigkeiten etc. Hilfreich für Behörden oder Lieferkettennachweise ⚠️ Warum liefert die Suche oft keine Ergebnisse?
Hier die häufigsten Ursachen und Tipps:
1. Falsche Schreibweise / fehlende Formatierung
- EUDAMED unterscheidet zwischen Groß-/Kleinschreibung
🔍Acura3000≠acura30002. Teilbegriffe funktionieren nicht
- Keine automatische „Enthält“-Suche → z. B.
Medifindet nichtMediPlus 5000
👉 Verwende möglichst den vollständigen Namen3. SRN falsch eingegeben
- SRNs müssen im exakten Format eingegeben werden (z. B.
DE-MF-000000xxx)4. Kein Zugriff auf fremde Einträge
- Du kannst nur Produkte/Akteure sehen, die mit deiner Organisation verknüpft sind
👉 Öffentlich sichtbare Daten kommen erst nach vollständiger Live-Schaltung von EUDAMED5. Veraltete/abgelaufene Sessions
- Die GUI hat ein Timeout, nach dem keine echten Ergebnisse mehr angezeigt werden – oft hilft: neu einloggen
Tipps für eine erfolgreiche Suche in der GUI:
Was du suchst Wie du es eingibst UDI-DI / GTIN Komplette Nummer exakt eingeben Basic UDI-DI Ohne Leerzeichen oder Sonderzeichen Gerätename / Modell Exakt wie im EUDAMED-Eintrag Hersteller / Importeur (SRN) Komplett mit Länderkürzel ( DE-MF-...)Status Optional: Suche nur nach aktiven Produkten ( Published)
Frage: Gibt es einen Upload über Excel? Wie können wir doppelte Datenpflege vermeiden – in Eudamed und intern?
❌ **Nein – aktuell unterstützt EUDAMED keinen direkten Excel-Upload.
Die EU-Kommission erlaubt nur:
Manuelle Eingabe über das GUI (Web-Portal)
Automatisierten Upload per XML-Datei nach dem offiziellen XSD-Schema (für Massenregistrierung oder M2M)
💡 Aber: Sie können mit Excelvorlage von Europe IT arbeiten
Wie vermeiden Sie doppelte Datenpflege?
Auch hier können wir unsere Excel Template empfehlen.
Frage: Empfehlen Sie bestimmte Tools für die Produktregistrierung von Produkten, bei denen wir die Herstellerrolle haben?
Ja, wir empfehlen die interne Lösung von Europe IT Consulting, basierend auf einer strukturierten Excel-Vorlage.
Gerne können Sie uns hierzu kontaktieren.
Frage: Wird Eudamed die Registrierung in lokalen Datenbanken ersetzen , sobald die Module voll funktionsfähig sind oder nicht?
Vermutlich ja. Wir nehmen an, dass sobald alle EUDAMED-Module vollständig funktionsfähig sind und offiziell im EU-Amtsblatt (OJEU) bekannt gemacht wurden, wird EUDAMED die nationalen Produktdatenbanken für MDR-/IVDR-konforme Produkte ersetzen.
Aber: In der Übergangsphase behalten viele Mitgliedstaaten ihre lokalen Systeme parallel bei.Was genau bedeutet das?
📌 Sobald EUDAMED vollständig in Betrieb ist (nach OJEU-Veröffentlichung):
Bereich Zuständigkeit UDI-/Device-Registrierung zentral in EUDAMED – ersetzt nationale Datenbanken Akteurregistrierung (SRN etc.) Nur noch über EUDAMED Marktüberwachung & Vigilanz EU-weit über EUDAMED sichtbar Verknüpfung von Herstellern, Importeuren, Bevollmächtigten Nur noch in EUDAMED gültig 📅 Aber bis dahin gilt: Übergangsphase = nationale Parallelstrukturen
Viele Behörden (z. B. BfArM in DE, FAMHP in BE) betreiben weiterhin eigene Datenbanken:
Beispiel Aktueller Stand Deutschland (BfArM) Nationale Registrierung für Altgeräte und gewisse IVDs weiterhin erforderlich Frankreich (ANSM) Verlangt teils zusätzliche nationale Einträge, z. B. zur Marktverfügbarkeit Italien (Ministerium) Fester Datentransfer von EUDAMED zu nationalem System geplant 📌 Was bedeutet das für euch in der Praxis?
Phase Was zu tun ist Jetzt (2025, freiwillige EUDAMED-Nutzung) EUDAMED nutzen = strategischer Vorteil (Vorbereitung, Audit-Sicherheit) Ab OJEU-Bekanntgabe (voraussichtlich 2025) 24 Monate Zeit für vollständige Migration Danach (verpflichtende Nutzung) Nur noch Registrierung in EUDAMED – nationale Systeme dienen evtl. nur noch zur Überwachung oder ergänzenden Zwecken
Frage: Welche lokalen Datenbanken sind weiterhin zu pflegen (z.B. Hersteller Ort – Deutschland – BfArM/DMIDS) und noch andere?
Auch wenn EUDAMED mittelfristig das zentrale System wird, gibt es aktuell (2025) mehrere nationale Datenbanken in der EU, die weiterhin gepflegt werden müssen, je nach Produkttyp, Standort und Marktanforderung.
Hier ist eine übersichtliche Liste der wichtigsten nationalen Datenbanken, die aktuell (Stand 2025) noch aktiv gepflegt werden müssen, insbesondere für Hersteller mit Sitz in der EU:🇩🇪 Deutschland – BfArM / DMIDS
Datenbank Zweck Pflicht? DMIDS (Deutsche Medizinprodukte-Informations- und Datenbank-System) Registrierung von Herstellern, Bevollmächtigten, System-/Prozedurpackern ✅ Ja, weiterhin Pflicht – bis EUDAMED offiziell verpflichtend ist DIMDI Altgeräte-Meldung (MDD/IVDD) Meldung von Legacy Devices ✅ Ja, falls noch MDD-/IVDD-Produkte im Verkehr sind UDI-Meldung ❌ Wird perspektivisch durch EUDAMED ersetzt 📌 Hersteller mit Sitz in DE müssen derzeit parallel EUDAMED und DMIDS pflegen, v. a. für Altgeräte oder nationale Marktüberwachung.
🇫🇷 Frankreich – ANSM
Datenbank Zweck Pflicht? Base de données des dispositifs médicaux (BDDM) Nationale Registrierung von Produkten, vor allem IVDs ✅ Ja ANSM-Formular für Marktverfügbarkeit Info an Behörden über Vermarktung in Frankreich ✅ Ja, auch bei EUDAMED-Eintrag 🇮🇹 Italien – Ministero della Salute
Datenbank Zweck Pflicht? Repertorio Dispositivi Medici (RDM) Pflichtregistrierung für Verkauf in Italien ✅ Ja – sogar bei EUDAMED-Registrierung zusätzlich Notifizierung über CND-Codes Italien nutzt CND statt EMDN – muss konvertiert werden ✅ Ja 🇪🇸 Spanien – AEMPS
Datenbank Zweck Pflicht? Registro de Productos Sanitarios (RPS) Registrierungspflicht vor Inverkehrbringen in Spanien ✅ Ja 🇧🇪 Belgien – FAMHP / AFMPS
Datenbank Zweck Pflicht? Webportal für Akteurregistrierung Registrierung des Herstellers, auch bei EUDAMED-Nutzung ✅ Ja Produktmeldung Noch notwendig für nationale Marktüberwachung ✅ Ja, temporär 🇦🇹 Österreich – BASG
Datenbank Zweck Pflicht? BASG MedProd-Portal Meldung von Produkten, insbesondere bei Vertrieb durch österreichische Stellen ✅ Ja, parallel zu EUDAMED 🔁 Was bedeutet das in der Praxis?
Wenn… Dann musst du… Ihr Firmensitz ist in Deutschland DMIDS-Eintrag + ggf. Altgeräte in DIMDI Sie vertreiben Produkte in Frankreich, Italien, Spanien, Belgien, Österreich Nationale Produktregistrierung zusätzlich zu EUDAMED durchführen Sie verkaufen nur in DACH-Raum Nur BfArM und BASG pflegen – bis EUDAMED aktiv wird