EUDAMED Questions and Answers
Contents
Actors & Roles in EUDAMED
Product Registration – Basics & Deadlines
- Deadlines: When will EUDAMED become mandatory?
- Registration requirement for Class I products
- UDI registration requirement for IVD products
- Registration requirement for IVDD stock products
Product Registration – Practice & Execution
- Register single product – step-by-step
- How the product registration in EUDAMED WORKS
- Edit Already Registered Products
- Changes to the product (name, trade name, etc.)
- Warnings & contraindications
- Change basic UDI-DI subsequently
Data structure & mandatory information
- Device model – what to enter?
- Mandatory data for product registration
- Market Information – Date Fields
- Country List: Applicable only to EU?
- “Originally placed on the market” – meaning
Grouping & Coding
GUI & System Functions
- Functions in the EUDAMED GUI
- Excel upload & avoid double data maintenance
- Tool recommendation for manufacturer role
National Systems vs. EUDAMED
Question: Can a company registered as an importer in EUDAMED link to a manufacturer registered in EUDAMED without sending a notification to the manufacturer and obtaining a permit from him?
No, this is not possible without the knowledge and active participation of the manufacturer .
In EUDAMED, the linking of actors – e.g. importer with manufacturer – is always a mutual processthat requires confirmation or approval by the other partner .In detail:
The importer can initiate the linkby indicating in his EUDAMED account (in the actor module) that he is affiliated with a specific manufacturer (stating its SRN).
The manufacturer must then explicitly accept this link request. Without the manufacturer’s approval, the link will not be active.
Question: The majority of our products fall under the manufacturer role, but there are some products that we bring to the market with the importer role. For the importer role, I would like to know how the registration will be done.
Registration as an importer in EUDAMED – step by step
1. Understand importer role (according to MDR, Article 13):
An importer is a company based within the EUthat brings a product onto the EU market from outside the EU – i.e., for example, first imports and distributes products from a non-EU manufacturer into the EU.
Important note:
Only necessary if you are importing products from a non-EU manufacturer.
If you are a manufacturer based in the EU and sell your own products, you are not an importer within the meaning of the MDR.
📝 How to register as an importer?
A. Step 1: Registration in the Actor module
Go to the EUDAMED Actor Registration Portal
Select “Importer” as the actor role.
Submit the actor registration to your national authority (in DE: BfArM, in FR: ANSM, etc.).
After approval, you will receive your SRN (Single Registration Number) as an importer.
📌 If you are already registered as a manufacturer, you must also carry out a second registration as an importer – with a separate role, but possibly the same company.
B. Step 2: Link to non-EU manufacturers
As soon as you have an SRN as an importer, you can link up with the respective non-EU manufacturers whose products you import into the EU.
The link is mutual – the manufacturer must agree.
This link is a prerequisite for the products in question to be correctly assigned in EUDAMED.
📌 Important obligations in the importer role (MDR Article 13):
Task: Description Ensuring MDR compliance The importer must check whether the product is CE marked, instructions for use are available, etc. Check UDI compliance Packaging must bear UDI label correctly Ensure product traceability Importer must document its supply chain Check UDI/Device entries in EUDAMED Imported products must be properly registered Cooperation with authorities Importer must be able to report recalls, incidents, etc.
🧠 Special feature: manufacturer & importer combination
For example, if you manufacture your own products (manufacturer role) and additionally import products from a non-EU manufacturer under your own SRN, then:
you need to register twice: once as a manufacturer, once as an importer
you must fulfil the importer obligations separately for the imported products
you may not register the same products as a manufacturer and importer at the same time – it must be clearly assigned
📋 Additional note:
For imported products, the non-EU manufacturer is responsible for product registration (device module).
The importer is linked in the system as an economic operator, not as the primary registrar.
Question: What is the clear demarcation of roles at EUDAMED?
Clear delineation of roles in EUDAMED – who does what?
There are 6 actor roles in EUDAMED, but for product registration and duties, these 4 are the most important:
Role: SRN required? Product registration possible? Typical tasks Manufacturer ✅ Yes ✅ Yes Registers products (UDI), manages device data, vigilance Authorised Representative ✅ Yes No (unless delegated by the manufacturer) Represents non-EU manufacturers, can take over administrative tasks Importer ✅ Yes ❌ No Links to manufacturer, assumes MDR obligations (e.g. label inspection, traceability) System/Procedure Pack Manufacturer ✅ Yes ✅ Yes Reports sets consisting of multiple products 🔍 1. The “Manufacturer” role
Has full control over product registration
Must register each Basic UDI-DI and UDI-DI
Can be both EU and non-EU manufacturers (non-EU mandated by EU authorised representatives)
Links products with economic operators (importers, distributors)
Responsible for vigilancereports, technical Documentation, intended purpose, etc.
|||UNTRANSLATED_CONTENT_START|||🔍 2. |||UNTRANSLATED_CONTENT_END|||The role of “importer”
Can only register if the registered office is within the EU
Does not register products itself – but must:
Linkingwith the manufacturer
Ensurethat the products it imports are registered in EUDAMED
Make your own company visible in the Economic Operator module
Importer must affix its SRN on the label (Art. 13 MDR)
📌 One and the same company can be both manufacturer and importer – but in EUDAMED these are two separate roles, each with its own SRN.
🔍 3. The Authorised Representative role
Necessary if the manufacturer is not based in the EU
Is linked to the non-EU manufacturer by a mandate declaration
Does not register products himself, unless he takes on this task on behalf of the manufacturer
👥 Linking the roles – example from practice
So if you:
Manufacturer is based in the EU → You register the products yourself (UDI data, etc.)
Simultaneously importer for products of a US manufacturer → You register separately as an importer, get your own SRN and link yourself with the US manufacturer
📄 In a nutshell: what does this mean for you?
If you… Then you have to… manufactures its own products in the EU Register as a manufacturer , register products yourself Products imported into the EU from third countries In addition, register as an importer and link to the manufacturer acts for non-EU manufacturers Either act as an authorised representative or ensure a contractual relationship
🎯 Conclusion:
EUDAMED
is structured with sharp roles – and each role has its own tasks, duties and its own SRN. Once a company takes on multiple roles, it needs clear internal processes and system mapping to avoid confusion or audit issues.
Question: Deadlines: The EU Commission has announced that the UDI registration module will soon be fully operational, with the deadline for registering the products being early next year. Is this deadline expected to be met and EUDAMED to be operational ON time?
Status: Q1/2025:
Yes, the EU Commission is expectedto officially confirm the full operation of the EUDAMED UDI/Device module in the first half of 2025 in the Official Journal of the EU (OJEU) . The deadline for mandatory product registration is then expected to be 24 months after this announcement .
👉 This means that the deadline will end at the beginning of 2027 at the earliest.
📅 What is the current status (March 2025)?
The UDI/device module is already technically fully functional and can be used voluntarily.
The EU Commission is currently conducting the latest interoperability tests (M2M) with economic operators and national authorities.
The official announcement in the EU Official Journal (OJEU) is still pending – it marks the “starting signal” for the 24-month transition period.
🧠 What does this mean for manufacturers & importers?
TOPIC: Meaning Voluntary use Already possible & recommended – helps with internal process security and data maintenance Mandatory use → Currently does not start a legal obligation until after OJEU announcement UDI Registration Obligation Required 24 months after OJEU release for all MDR products Recommendation: Start registration now – to avoid bottlenecks and errors 📌 Practical tip:
Many companies underestimate the effort required to maintain UDI data, especially for large portfolios. If you wait until the commitment, you run the risk of not making the effort in the transitional period – or having problems in audits.
💬 Conclusion:
The deadline is expected to be published in the Official Journal – presumably in the course of 2025.
The registration obligation will apply from then on + 24 months, so presumably from the beginning of 2027.
Technically, EUDAMED is ready – companies should prepare now, even if the obligation does not yet apply.
Question: What are the deadlines for registering Class I products?
The registration obligation for Class I products in EUDAMED only begins when the EU Commission officially publishes the full functionality of EUDAMED in the Official Journal (OJEU). From this point onwards, a transition period of 6 months applies.
📅 In concrete terms, this means:
Step : Date OJEU publication (full EUDAMED function) ⚠️ Pending (expected 2025) Start of product registration obligation (Class I) 📆 6 months after OJEU publication Registration must be completed 📆 Expected in early/mid-2026, depending on release date
Question: do I already have to use EUDAMED for UDI labelling (according to IVDR) or when is it necessary?
No, there is currently no legal obligation to register UDI data in EUDAMED – not even under IVDR.BUT:
The EU Commission is expected to announce the full functionality of EUDAMED (including UDI module) in the Official Journal of the EU (OJEU) in the course of 2025.👉 From this point onwards, there is a 24-month transition period.This means:UDI registration in EUDAMED will become mandatory from 2027 at the earliest – also for IVD products.
Question: Are existing products according to IVDD also to be deposited there?
No, existing products that have still been placed on the market under the IVDD do not currently have to be registered in EUDAMED.
The UDI registration obligation only applies to IVDR-compliant products.Transition Period & Exceptions
🧾 What are “Inventory Products”?
These are products that have been certified according to IVDD and may continue to be placed on the market in accordance with the transitional periods of IVDR Article 110 – e.g., on the basis of an old Notified Body certificate. Depending on the class, such products may continue to be marketed until May 2026 or May 2027 at the latest , without already having to be IVDR-compliant.
❗ The following applies to these products:
You don’t need a UDI tag yet
You do not need to be registeredin EUDAMED
There are no new obligations in EUDAMED – neither for the device module nor for vigilance
🚦 When do IVD products need to be registered in EUDAMED?
Product Type Registration required in EUDAMED? New IVDR Compliant Product ✅ Yes, after the start of the UDI obligation (expected from 2027) Inventory product according to IVDD ❌ No, as long as it is resold under the transitional rule Converted product (IVDR recertification) ✅ Yes
Question: How do you register a single Class I product?
Registering an individual product in EUDAMED – here’s how:
Open UDI/Devices module
Select“Register new UDI/Device”
Gothrough the wizard step by step (5 steps):
Step 1: Basic UDI-DI, Device Name, EMDN, Risk Class, Purpose
Step 2: UDI-DI (GTIN), packaging, sterility
Step 3: Economic Operators (manufacturer, authorised representative/importer if applicable)
Step 4: Market availability (“from”date, “to”date if applicable)
Step 5: Validate & Submit
Check status: Product is displayed as “Submitted” or “Published”
Question: How exactly do you make product entries in the EUDAMED?
Requirement:
Question: How can registered products be subsequently processed?
✅ To edit an already registered product in EUDAMED:
Log in to EUDAMED
👉 HTTPS://WEBGATE.EC.EUROPA.EU/EUDAMEDOpen UDI/Devices module
Search product via UDI-DI or Basic UDI-DI
Click on “Actions” “Update→” to the right of the entry
Go through the wizard and change the desired fields
Validate and then submit – the update will be submitted and processed
📌 Important:
You can only edit productsthat are linked to your SRN as a manufacturer
Some fields (e.g. Basic UDI-DI) cannot be changed – create a new product if necessary
Question: The cooperation between the tasks (e.g. changing the product name or adding a trade name) and the associated activities Actions with the GUI of EUDAMED as well as the meaningful order of the activities/actions in the GUI.
Typical change examples in the GUI
Änderung Affected field (GUI) EUDAMED module Action Required Change product name “Device Name” UDI/Device Module Submission Update Add trade name “Brand Name” UDI/Device Module Submission Update Modify model number “Model” UDI/Device Module Submission Update Adjust Intended Purpose Intended use UDI/Device Module Submission Update Add packaging information “Packaging” UDI/Device Module Submission Update Change sterility/label data “Device Characteristics” UDI/Device Module Submission Update 🖥️ Procedure in the GUI – recommended sequence of steps
🎯 Scenario: You want to change/add product name AND trade name
🔄 1. Login & Select Product
Go to the UDI/Devicemodule
Search for the relevant UDI-DI (GTIN)
Click on “Actions” → “Update”
✍️ 2. Step-by-step adjustment in the wizard
The GUI is divided into steps – the following fields are relevant:
Step 1: Basic UDI-DI & Device Information
Here you can customize Device Name (product name) and Brand Name (trade name)
You can also add several trade names here (e.g. for multilingual markets)
Step 2: Device Characteristics
Check if the name change affects other data (e.g. label language, packaging)
Step 3–5: Review and Submit
After the last page, you can check your entries
Click on “Validate” → the system checks if all mandatory fields are correct
- Then click on “Submit”
🔐 3. After Submit
- The status of the change is displayed as “Under Review” or “Updated”
- Some changes appear immediately, others only after approval by the competent authority (depending on the field and national implementation)
⚠️ Important Notes on Sequence & Logic
- Device Name vs. Brand Name:
- Device Name = technical product name
- Brand Name = commercial brand on the market (e.g. “MediPlus®”)
- Brand name can be multiple, device name not → brand name so choose wisely
- Changes to product name or brand name can have repercussions on label, IFU or technical Have documentation – so vote internally beforehand
- You can combine multiple changes in one update submission – this is more efficientthan submitting everything individually
Question: Could you please provide examples of critical warnings or contraindications (step 4: UDI-DI characteristics)?
In this field, the manufacturer should indicate relevant warnings or contraindications to the product, if any. The goal is to make patient and user safety directly visible.
✅ Examples of typical critical warnings:
“Not suitable for use in patients with implanted defibrillator.”
“Do not use near magnetic resonance imaging (MRI) scanners.”
“Only use by trained specialists.”
“Sterile – do not use if packaging is damaged.”
✅ Examples of contraindications:
“Do not use in case of known allergy to latex.”
NOT SUITABLE FOR CHILDREN UNDER 3 YEARS.
“Do not use in case of open wounds or skin diseases in the application area.”
“Do not use in pregnant or breastfeeding women.”
⚠️ Important:
If there are no warnings or contraindications, the field can be filled with “None known” – but never leave empty.
The input is in free text, so you should formulate it concisely but concisely.
Question: I would like to know the best way to correct or change a Basic UDI-DI in EUDAMED. I tried to create a new entry with a different Basic UDI-DI for a previously registered product. However, I receive an error message (step 3: UDI-DI identification information) because the UDI-DI code/GTIN of the device has already been used for my previous entry.
The Basic UDI-DI is the superordinate identification feature of a product group (e.g. same purpose, risk class, essential design).
It is permanently linked to the product UDI-DI (GTIN) and cannot simply be exchanged after submissionif an entry with this GTIN already exists.🔒 Why does the error message appear?If you try to register a product with a new Basic UDI-DI, but use a UDI-DI (GTIN) that has already been submitted with another Basic UDI-DI, EUDAMED detects this as an inconsistency and blocks the process:
Error: The UDI DI code has already been used for another basic UDI DI.
Possible solutions:
1. Updateexisting listing, do not resubmit
If the UDI-DI/GTIN is already registered, you can only go through a change to the existing entry – for example, through an update submission (via Web Interface or M2M/XML), not through a new registration.
👉 But beware: the Basic UDI-DI itself cannot be changed on an existing device.
2. If the Basic UDI-DI was really wrong (e.g. typos)
Because then there are only two possibilities:
Log off device (deactivate/delete) and re-register with correct basic UDI-DI and UDI-DI
Or (if the device has not yet been actively marketed): generate a new UDI-DI (new GTIN) and re-register with the correct Basic UDI-DI
📌 Important: The link UDI-DI ↔ Basic UDI-DI is permanent. The same GTIN cannot exist with two different Basic UDI-DIs.
3. If multiple similar products are to be registered:
Then they should each receive their own GTIN – even if they differ in details – and be registered via their own basic UDI-DIs, if justified (different purpose, risk class, etc.).
Question: Could you please describe what correct information needs to be entered in the ‘Device model’ field (Step 1: Basic UDI-DI information)? I have noticed that manufacturers do not use this field consistently in EUDAMED.
Definition according to MDCG 2018-1:
The device model describes the technical or commercial designation under which the medical device is identifiably marketed or used. It helps to distinguish products within a basic UDI-DI group.✅ The correct answer would therefore be:
A technical model name that is mentioned in the label, in the instructions for use or in the product documentation.
Or an item number-related designationif no model number is available.
❌ The following are not suitable:
Product descriptions (“Sterile disposable catheter set for children”)
Purposes (“For the treatment of XYZ”)
Free texts with keywords or variant combinations
🔍 Examples of correct device model entries:
Product Type Device Model Pacemaker Model ACURA 3000 Disposable surgical mask Type 2R – MaskCare Blue Dental implant IMPL-24 Titanium Software (SaMD) Diagware v1.4.2 Blood pressure monitor BP Monitor Pro M120
Question: Which data elements are mandatory for product registration?
Mandatory fields for product registration in EUDAMED (UDI-DI level)
Sorted by category here:📘 A. Basic Device Information (Step 1 in GUI)
Field Duty? Comment Basic UDI-DI ✅ Yes Vom Hersteller selbst vergeben (z. B. über GS1) “Device Name” ✅ Yes Technical name (no marketing text) “Brand Name” ❌ No Only if used by the market EMDN Code ✅ Yes EU nomenclature code (min. 4-digit) Regulatory Status (MDR/IVDR) ✅ Yes Indicates whether MDR or IVDR Risk Class ✅ Yes I, IIa, IIb, III (MDR) or A–D (IVDR) Intended Purpose ✅ Yes Intended purpose in plain text Notified Body Information ✅ Yes, if applicable Only for products with NB certificate 📦 B. UDI-DI & Packaging (Step 2 in GUI)
Field Duty? Comment UDI-DI (GTIN or similar) ✅ Yes Main identifier on the packaging Primary Packaging Type ✅ Yes E.g. individual packaging, bulk etc. Sterility Information ✅ If product is sterile Otherwise, it may remain empty Storage & Shelf Life ❌ Optional Only in case of temperature/decay dependency Critical Warnings / Contraindications ❌ Optional If available, enter 👥 C. Economic Operators (Step 3 in GUI)
Field Duty? Comment Manufacturer (SRN) ✅ Yes Automatic if you are a manufacturer yourself – the authorized representative – ✅ If you are a non-EU manufacturer Must have valid SRN Importer ❌ Optional Nur falls vorhanden – sonst leer lassen 🌍 D. Market Information (Step 4 in GUI)
Field Duty? Comment “From”date (Date from) ✅ Yes Market Availability Start Date “To”date (Date to) ❌ No Only in case of discontinuation/market withdrawal 📎 E. Attachments (Step 5 in GUI)
Field Duty? Comment Label / IFU Upload ❌ Optional May be required during audits, but not mandatory Technical Documentation ❌ Not directly in EUDAMED Required only for government request
Question: Request clarification regarding the “Market Information” section in EUDAMED, specifically regarding the “from (YYYY-MM-DD)” and “to (YYYY-MM-DD)” date fields. What exactly does this data stand for or do they have to be filled in?
This depends on the legislation under which you register your product. If you select the old legislation (MDD, IVDD, AIMDD), you must enter at least the “valid until”date (optionally you can also specify the start date). But only the “to”date is mandatory. If, on the other hand, you select the new regulation such as “MDR” or “IVDR”, you do not have to specify a date at all.
What do the “from (YYYY-MM-DD)” and “to (YYYY-MM-DD)” fields in the “Market Information” section stand for?
Field Meaning “from”date ( Date from which the device is/was made available on the market)The date from which the product was first made available on an EU market (in accordance with Article 2 No. 27 MDR/IVDR: “Placing on the market”) “to”date ( Date until which the device is/was made available on the market)The date by which the product is or was active on the market – e.g. in case of withdrawal from the market, product discontinuation, deactivation 🧠 What does this mean in practice?
The “from”date is typically the launch date in the EU
The “to”date is only setif the product is no longer actively marketed or has a fixed expiry date
📌 Example:
A product was launched on 01/01/2023 and is still available:
→ “from” = 2023-01-01→
“to” = leave blank (not required)⚠️ Are the fields mandatory?
▶️ Current status according to the EU Commission (XSD v2.0 & GUI specification):
Field |||UNTRANSLATED_CONTENT_START|||Pflichtfeld?|||UNTRANSLATED_CONTENT_END||| “from”date ✅ Yes, mandatory “to”date ❌ No, only required in case of discontinuation or end of market
Question: Section 2 “List of all countries where the product is or will be available”, does this only apply to EU countries?
Yes, only EU countries can be selected in this section.
Question: Indication that a country must be defined as “originally placed on the market”, what is meant by this? The country in which the medical device (UDI dataset) was first placed on the market?
Does this information only refer to a first EU member state, or can third countries outside the EU also be taken into account in this field?
EUDAMED refers exclusively to the EU market. Countries outside the EU are not part of this system. Manufacturers who wish to market their products in non-EU countries must meet the respective regulatory requirements of these countries separately.
Question: Is it advisable that products are merged and registered according to Basic UDIs? (clusters or separately), e.g. hoses for insufflators – keyword: is it possible to group products to minimise workload?
Yes, a meaningful grouping (“clustering”) of products under a common basic UDI-DI is not only permissible, but also expressly provided for – as long as certain criteria are met. This significantly reduces the registration and maintenance effort in EUDAMED.
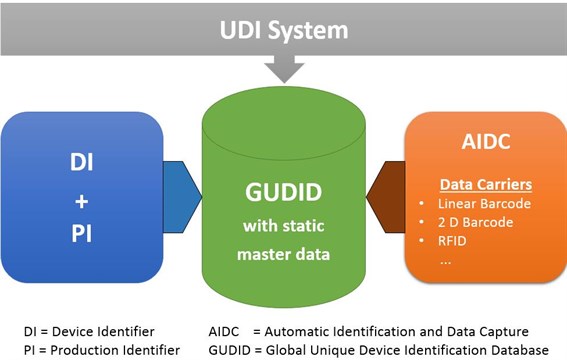
📘 What is Basic UDI-DI and what is it for?
The Basic UDI-DI is:
the primary key for the product group
the connection to the declaration of conformity, certification, technical Documentation
not printed on the packaging, but only in EUDAMED & Doku
📦 When can products be grouped under a common basic UDI-DI?
According to MDCG 2018-1 (and EU Commission):
✅ The products must:
Criterion: Meaning Same purpose They serve the same clinical or diagnostic purpose Same risk class e.g. all class IIa Similar basic construction / design e.g. hoses made of the same material, same connection Part of the same technical Documentation They are evaluated in a file, e.g. together in a DoC 🧪 Example: insufflator hoses
These can be easily grouped if:
they have the same function (e.g. connecting insufflator to patient)
differ only in length, colour or packaging unit
the same manufacturer produces them and a common technical document is available
👉 Then it is advisable to cluster these variants under a Basic UDI-DI and register several UDI-DI (GTINs) under this base.
🎯 Advantages of clustering (grouping):
Advantage Description 🔁 Less effort Only one base-level registration with multiple UDI-DIs 🧾 Unified Documentation Only a declaration of conformity & technical file required 🔍 Better overview You can manage your variants in a more structured way in EUDAMED 📈 Scalability New variants can be added more easily later ⚠️ But beware: When not to cluster:
Do not cluster at… Reason? Different intended purpose e.g. hose for CO₂ vs. for other gases Different risk classes e.g. Class I vs. Class IIa Different regulatory assessments e.g. one variant sterile, the other not Different certificates or notified bodies → separate basic UDI-DIs required
Question: May we use GS1’s basic UDI-Dis for legacy devices or do we have to have them generated by Eudamed? This means that the latter must be linked to the new ones?
Yes, you may also use GS1-generated Basic UDI-DIs for legacy devices – automatic generation by EUDAMED is not necessary.👉 You must assign the Basic UDI-DI yourself (e.g. Via GS1) – also for legacy products – if you want to register them in EUDAMED (voluntary or later mandatory).
Question: Is there a way to generate new EMDN codes if a new product with a new technology cannot be found in the existing codes?
No, manufacturers cannot generate their own EMDN codes. New codes can only be added by the EU Commission – upon request or via defined channels.What to do if there is no matching EMDN code?
Select the nearest EMDN code
You are required to choose the best applicable existing code
For example, if your product is an innovative portable dialysis machine, but there are only codes for classic dialysis machines – then take the closest one
As a rule, 4-digit or 6-digit codes (level 2 or 3) are sufficient for product registration
📌 This selection must be well documented – also for audits (e.g. why you chose this code).
Question: What else can you do with the GUI, e.g. you can search for specific products, product classes, manufacturers, importers, etc. Search often returns empty answers.
Typical functions in the UDI/Device module:
Role Description Comment Search 🔍 products By UDI-DI (GTIN), Basic UDI-DI, Device Name, Brand Name Often case-sensitive and very restrictive View 🗂️ product details All details about the registered product incl. Packaging, label, intended use, etc. Only if the search hit is correct Change ✏️ product (update submission) Change or supplement product information Only for products that originate from your own SRN Deactivate 🗑️ product (Deactivate) Withdraw or withdraw product from the market Only possible by the registering manufacturer Register ⬆️ new product With complete wizard over 5 steps Basic UDI-DI must be correct - Check product status „Submitted“, „Under Review“, „Published“ etc. Important overview of workflow status Typical functions in the Actor module:
Role Description Comment 🔍 Search for actors by SRN, company name, role (manufacturer, importer, etc.) often only works with exact spelling 🔗 Linking with other actors e.g. manufacturer with importer or authorized representative mutual confirmation required 🧾 Show actor details Address, roles, responsibilities, etc. Helpful for authorities or supply chain verification ⚠️Why does the search often not return any results?
Here are the most common causes and tips:1. Incorrect spelling / missing formatting
EUDAMED is case-sensitive
🔍 Acura3000 ≠ acura30002. Partial terms don’t work
No automatic “contains” search → e.g., Medi doesn’t find MediPlus 5000
👉 Use the full name if possible3. SRN entered incorrectly
SRNs must be entered in the exact format (e.g., DE-MF-000000xxx)
4. No access to external entries
You can only see products/actors linked to your organization
👉 Publicly visible data will only be available after EUDAMED is fully live5. Outdated/expired sessions
- The GUI has a timeout after which no real results are displayed – logging in again often helps.
Tips for a successful search in the GUI:
What you are looking for How to enter it UDI-DI / GTIN Enter the complete number exactly Basic UDI-DI No spaces or special characters Device name / Model Exactly as in the EUDAMED entry Manufacturer / Importeur (SRN) Complete with country code (DE-MF-…) Status Optional: Search only for active products (Published)
Question: Is there an upload via Excel? How can we avoid duplicate data maintenance—in Eudamed and internally?
❌ No – EUDAMED currently does not support direct Excel upload.
The EU Commission only allows:
Manual entry via the GUI (web portal)
Automated upload via XML file according to the official XSD schema (for mass registration or M2M)
💡 But: You can work with Europe IT’s Excel template.
How do you avoid duplicate data maintenance?
We can also recommend our Excel template here.
Question: Do you recommend any specific tools for product registration of products where we have the manufacturer role?
Yes, we recommend Europe IT Consulting’s internal solution, based on a structured Excel template. Feel free to contact us about this.
Question: Will Eudamed replace registration in local databases once the modules are fully operational or not?
Probably yes. We expect that once all EUDAMED modules are fully operational and officially published in the Official Journal of the European Union (OJEU), EUDAMED will replace the national product databases for MDR/IVDR-compliant devices.
However, during the transition period, many Member States will maintain their local systems in parallel.What exactly does that mean?
📌 Once EUDAMED is fully operational (after OJEU publication):
Field Responsibility UDI-/Device-Registration centrally in EUDAMED – replaces national databases Actor Registration (SRN) Only via EUDAMED Market surveillance & vigilance Visible throughout the EU via EUDAMED Linking manufacturers, importers, authorized representatives Only valid in EUDAMED 📅 But until then: transition phase = national parallel structures
Many authorities (e.g. BfArM in Germany, FAMHP in Belgium) continue to operate their own databases:
Example Aktueller Stand Germany (BfArM) National registration for waste equipment and certain IVDs still required France (ANSM) Requires some additional national entries, e.g., on market availability Italy (Ministry) Fixed data transfer from EUDAMED to national system planned 📌 What does this mean for you in practice?
Phase What to do Now (2025, voluntary EUDAMED use) Using EUDAMED = strategic advantage (preparation, audit security) From OJEU announcement (expected 2025) 24 months for complete migration Afterwards (mandatory use) Registration in EUDAMED only – national systems may only serve for monitoring or supplementary purposes
Question: Which local databases still need to be maintained (e.g. manufacturer location – Germany – BfArM/DMIDS) and others?
Even if EUDAMED becomes the central system in the medium term, there are currently (2025) several national databases in the EU that still need to be maintained, depending on the product type, location, and market requirements.
Here is a clear list of the most important national databases that currently (as of 2025) still need to be actively maintained, especially for manufacturers based in the EU:🇩🇪 Germany – BfArM / DMIDS
Database Aim Duty? DMIDS (German Medical Device Information and Database System) Registration of manufacturers, authorized representatives, system/procedure packers ✅ Yes, still mandatory – until EUDAMED is officially mandatory DIMDI Old Device Report (MDD/IVDD) Reporting legacy devices ✅ Yes, if MDD/IVDD products are still on the market UDI Notice ❌ Will be replaced by EUDAMED in the future 📌 Manufacturers based in Germany currently have to maintain EUDAMED and DMIDS in parallel, especially for waste equipment or national market surveillance.
🇫🇷 France – ANSM
Database Aim Duty? Base de données des dispositifs médicaux (BDDM) National registration of products, especially IVDs ✅ Yes ANSM form for market availability Information to authorities about marketing in France ✅ Yes, even with EUDAMED entry 🇮🇹 Italy – Ministero della Salute
Database Aim Duty? Repertorio Dispositivi Medici (RDM) Mandatory registration for sale in Italy ✅ Yes – even with EUDAMED registration in addition Notification of CND codes Italy uses CND instead of EMDN – must be converted ✅ Yes 🇪🇸 Spain – AEMPS
Database Aim Duty? Registro de Productos Sanitarios (RPS) Registration requirement before placing on the market in Spain ✅ Yes 🇧🇪 Belgium – FAMHP / AFMPS
Database Aim Duty? Web portal for actor registration Registration of the manufacturer, also for EUDAMED use ✅ Yes Product notification Still necessary for national market surveillance ✅ Yes, temporaly 🇦🇹 Austria – BASG
Database Aim Duty? BASG MedProd-Portal Reporting of products, especially when distributed by Austrian authorities ✅ Yes, in parallel with EUDAMED 🔁 What does this mean in practice?
If… Then you have to… your headquarters are in Germany DMIDS entry + old devices in DIMDI if applicable you distribute products in France, Italy, Spain, Belgium, Austria conduct national product registration in addition to EUDAMED you only sell in the DACH region only BfArM and BASG maintain – until EUDAMED becomes active